

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Huntington’s disease (HD) is an inherited disorder that results in selective neuronal death in the striatum. The primary cause of HD is a polyglutamine expansion in the huntingtin gene (HTT) that is induced by a repeated cytosine-adenineguanine (CAG) codon [14]. Proteolysis of an N-terminal fragment of mutant HTT isoforms produces aggregates in the nuclei and cytoplasm of spiny projection neurons in the striatum, followed by selective degeneration of the affected neurons [32]. Accordingly, various clinical symptoms develop gradually, including abnormal movements and cognitive dysfunction. Symptoms usually begin between 30 and 50 years of age, and patients with HD survive for an average of 10–20 years after symptom onset before death [10,13]. Despite our knowledge of the HTT gene and its encoded protein, the exact pathogenesis of HD remains largely unknown, hindering effective treatment. Medical treatments relieve some symptoms, but unfortunately, no cure is available for HD [13].
A proper animal model for HD is essential to determine the exact mechanism of HD and to develop new therapeutic modalities, such as cell therapy. Thus, some investigators have developed animal models, including a drug-induced rat model using quinolinic acid and knock-in and transgenic mouse models [8,27,31,33,34]. In addition, in vivo genetic rat models induced by lentiviral-mediated expression of mutant HTT in the striatum have been introduced [6,20,26]. In the present study, we used adeno-associated virus (AAV) to deliver mutant HTT into the rat striatum. AAV has some advantages, including the induction of a weak host immune response, a lack of toxicity in humans, and long-term stability after insertion into host cells compared to lentiviral vectors [5,18]. Previous studies reported the use of AAV for the delivery of 82Q [12], 97Q [29], or 138Q [3] into the mouse or rat striatum to generate an HD model; however, these reports only focused on the neuropathological characteristics of HD. To the best of our knowledge, no optimum genetic rat Huntington model using an adeno-associated virus (AAV-2) vector has been reported to date
We aimed to test whether infection of two doses [2×1012 genome copies (GC)/mL (×1, low dose) or 2×1013 GC/mL (×10, high dose)] of a customized AAV2 vector with 82 polyglutamines (AAV2-82Q) into the rat striatum altered optimal Huntington behavior and HTT protein expression. Therefore, we investigated changes in behavioral, neuropathological characteristics following direct infection of AAV2-82Q into the caudate putamen of the dorsal striatum in the rat brain to develop a more stable and useful animal model and to overcome the shortcomings of previous animal models.
MATERIALS AND METHODS
Animals
All protocols using animals were approved by the Institutional Animal Care and Use Committee at Chungbuk National University (CBNUR-1013-16, CBNUR-1310-19). Forty female Sprague Dawley rats weighing 250–300 g (Daehanbiolink, Eumseong, Korea) were used in this randomized, double-blind, controlled animal trial. The animals were assigned a group designation on the same day. The animals were housed in a laboratory with a controlled temperature that was maintained on a 12-hour light-dark cycle. Food and water were provided ad libitum.
AAV2 vector
The components of the AAV2 vector are illustrated in Supplementary Fig. 1. AAV2 is a ssDNA vector that is approximately 4.5 kb in length. The viral genome comprises two open reading frames and inverted terminal repeats at each end of the DNA strand. We customized pAAV2-CMV-HTT171-82QWPRE (concentration, 2.0×1013 GC/mL) from the SIRION Foundation (SIRION, Martinsried, Germany). The human huntingtin cDNA encoding for the first 171 amino acids of the protein along with 82 CAG repeats was cloned into transfer vectors with a cytomegalovirus (CMV) internal promoter using a restricted enzyme. The viral preparation was used to infect human 293T cells, purified and concentrated on a heparin affinity column. The final titer was determined by quantitative RT-PCR.
Experimental design
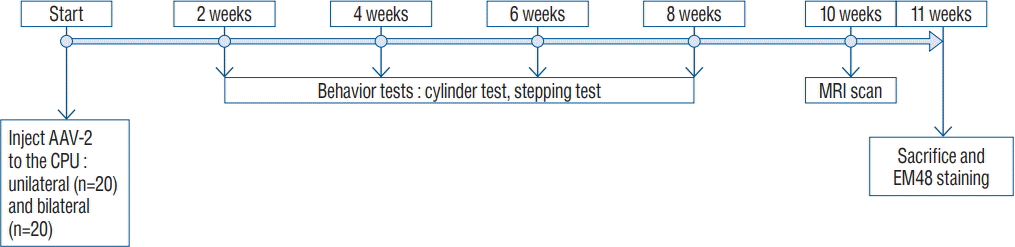
A schematic of the experimental schedule is illustrated in Fig. 1. We divided 40 rats into two experimental groups (20 rats each in the unilateral injection and bilateral injection groups). Each group included four subgroups composed of two AAV-82Q injection groups : 2×1012 GC/mL (×1, low dose) group, 2×1013 GC/mL (×10, high dose), and two AAV-GFP control groups (Table 1). We performed the stepping and cylinder tests 2, 4, 6, and 8 weeks after the AAV2-82Q injection. At week 10, immunohistochemical staining was performed for detection of neuronal dysfunction and HD protein aggregation.
Injection of the AAV2-82Q vector into the striatum
The stereotaxic viral vector injections (RWD Lifescience®, San Diego, CA, USA) were performed under anesthesia with a combination of 15 mg/kg of tiletamine/zolazepam (Zoletil50®; Virbac Lab., Carros, France) and 9 mg/kg of xylazine (Rompun ®; Bayer, Seoul, Korea) in saline to reduce handling-induced stress. The animals in each respective subgroup of the unilateral injection group received a 2-μL phosphate-buffered saline (PBS), 2×109 GC (2 μL of 10-fold-diluted AAV2 in PBS), or 2×1010 GC (2 μL of AAV2) injection using a microsyringe (Hamilton, NV, USA) into the right caudate putamen at the following coordinates : 0.43 mm posterior from the bregma, 3.0 mm lateral to the midline, and 5.4 mm ventral from the surface of the brain. When the microsyringe reached the target coordinates, we injected PBS or the AAV2-82Q vector at a rate of 0.2 μL/min using an automated micropump (Harvard Apparatus, Holliston, MA, USA) for 10 minutes. After the injection, we waited 5 minutes for absorption and then slowly removed the microsyringe. The skin was closed using a 4-0 nylon suture (Covidien, Seoul, Korea). The animals in the bilateral injection group received PBS or the AAV2-82Q vector in the bilateral caudate putamen using the same method.
Behavioral tests
We performed two behavioral tests (the stepping and cylinder tests) 2, 4, 6, and 8 weeks after AAV2-82Q vector injection to confirm changes in locomotion according to the injection site. For the stepping test, we used a treadmill apparatus (Panlabs1, Cornella, Spain) to assess dyskinesia in rats. After rats adapted to the treadmill by walking on the treadmill at a speed of 18 cm/s for 10 minutes and then resting for 5 minutes, the examiner firmly grasped and raised the hind limbs and truncus to shift the rat’s weight to the forelimbs. We started the treadmill again at a speed of 18 cm/s and recorded the number of steps for 20 seconds19). Another test used to assess the motor function of the rats was the cylinder test. For this test, we placed the rats into a transparent glass cylinder with a diameter of 20 cm and a height of 38 cm. The number of times each paw was placed on the cylinder wall during the experiment was recorded (a total of 20 paw placements per trial). We calculated the asymmetric score as (the affected paw+both paws/2) divided by (affected paw+contralateral paw+both paws) [11].
Histopathology
Eleven weeks after virus injection, the animals were anesthetized and perfused with 4% paraformaldehyde (Biosesang, Seongnam, Korea). Then, the brain was removed and immediately immersed in the same fixative for postfixation. After 24 hours, the animal brains were incubated in 30% sucrose for immunostaining. The staining process was performed as previously described [4,7].
Briefly, the rat brain was dehydrated in an ethanol series and embedded in paraffin. Serial coronal sections of the striatum (10-μm thick) were cut with a cryostat and mounted onto slides. Sections were blocked with normal goat serum for 1 hour at room temperature and then incubated overnight with primary antibodies against mouse anti-HTT (1 : 500, clone 1HU-4C8, catalog mAb2166; Merck Millipore, Darmstadt, Germany), mouse anti-HTT protein (1 : 200, clone mEM48; Merck Millipore), mouse anti-ionized calcium-binding adapter molecule 1 (Iba-1) (1 : 200; Abcam, Cambridge, MA, USA), mouse anti-glial fibrillary acidic protein (GFAP) (1 : 200; Abcam), rabbit anti-oligodendrocyte transcription factor (Olig2) (1 : 200; Abcam), goat anti-NeuN (1 : 200; Abcam) and rabbit anti-cleaved caspase 3 (1 : 200; Abcam). Sections were incubated with the corresponding secondary antibody (Vectastain Elite ABC-HRP Universal Kit; Vector Laboratory, Burlingame, CA, USA) for 1 hour. Then, sections were stained with DAB (Vector Laboratory). The nuclei were stained with hematoxylin. Finally, sections were dehydrated in an ethanol series, mounted with coverslips, dried and examined under a microscope.
Three to five slides per animal were investigated under microscope and the striatal area was identified manually using a rat atlas to count the number of immunohistochemically stained cells. We captured images of the injected area under a ×20 objective lens and counted the EM48-positive cells using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
SPSS version 20.0 (IBM, Armonk, NY, USA) was used for the statistical analysis. We used one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis to compare differences among the four subgroups and repeated measures ANOVA with a post hoc analysis to compare repeatedly measured data within each subgroup. The Mann-Whitney U test was used to determine the significance of differences in EM48-positive cell counts between two groups. p values <0.05 were considered significant differences.
RESULTS
Changes in rat behaviors
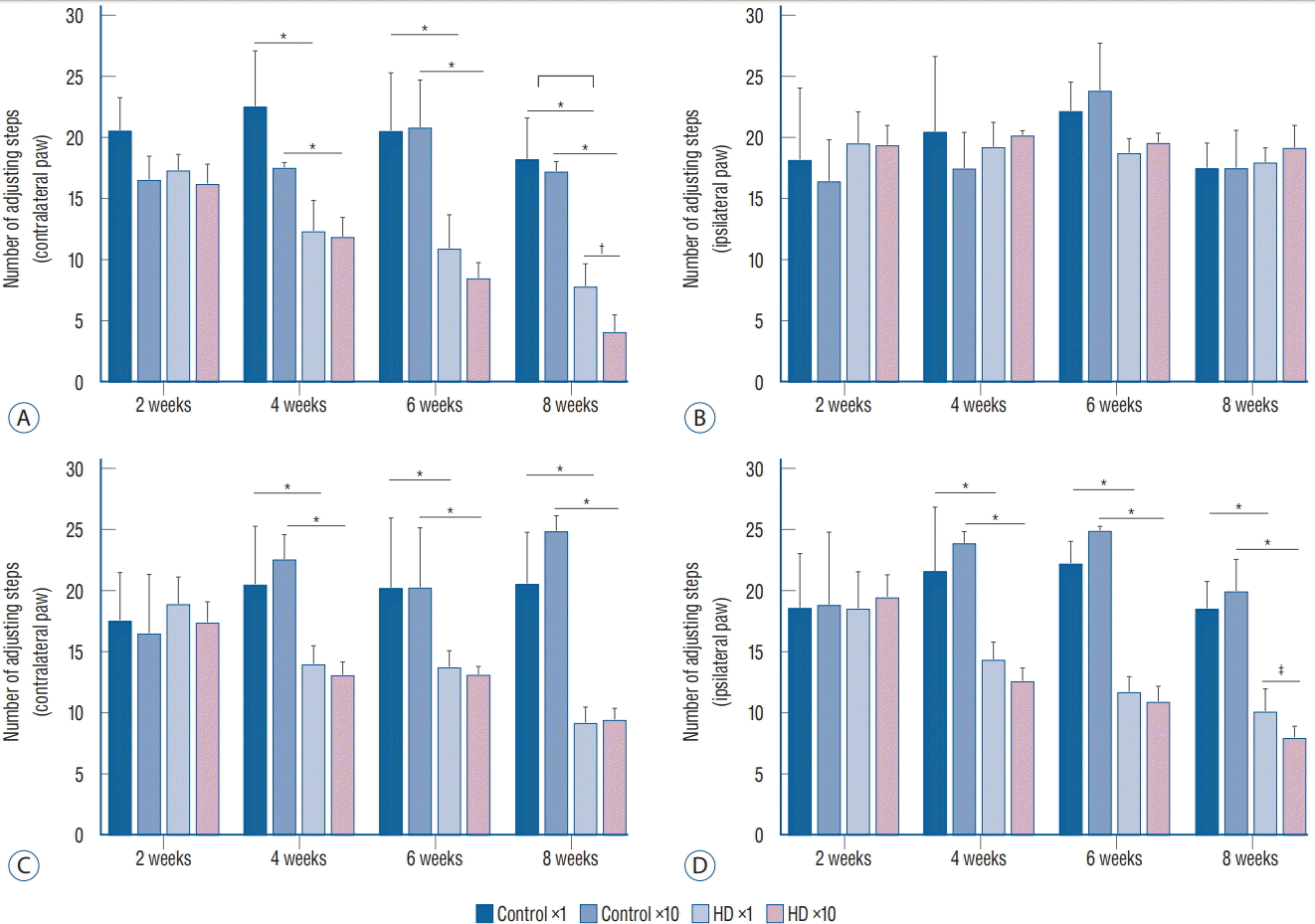
We conducted two behavioral tests at weeks 2, 4, 6, and 8 after the injection of the AAV2-82Q vector. In the stepping test, the number of steps of the affected paw (contralateral forelimb) was significantly decreased in the ×1 and ×10 subgroups of the unilateral model at 2 weeks compared to the control subgroups, whereas the number of steps taken by the unaffected paws plateaued (Fig. 2A and B). In the bilateral model, the numbers of steps of both the right and left paws were decreased in the ×1 and ×10 subgroups compared to the control subgroups (Fig. 2C and D). A significant difference in the stepping test results for the contralateral forelimb was observed between the unilateral and bilateral ×1 and ×10 subgroups at week 8 after injection (Fig. 2A), but no significant differences were observed between the ipsilateral forelimbs in the bilateral ×1 and ×10 subgroups (Fig. 2C and D). Repeated measures ANOVA with Tukey’s post hoc test revealed a significant decrease in the number of steps taken by the affected paw over time in both the ×1 and ×10 subgroups.
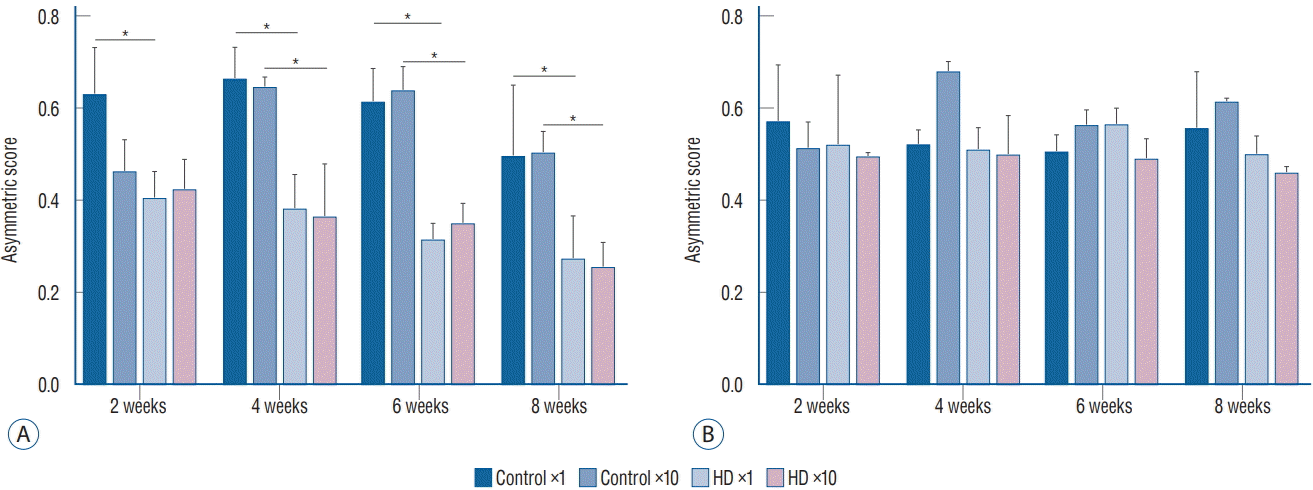
In the cylinder test, we compared the asymmetry scores among the three subgroups (control vs. ×1 vs. ×10 groups) in each unilateral and bilateral group. The asymmetry scores of the unilateral ×1 and ×10 subgroups were significantly decreased at 4 weeks after virus injection compared to the control subgroup. In the bilateral group, the asymmetry score did not differ between the control and injection groups. Additionally, no significant difference was observed in the asymmetry score between the ×1 and ×10 subgroups in the unilateral group (Fig. 3).
Histopathological changes and EM48 aggregation
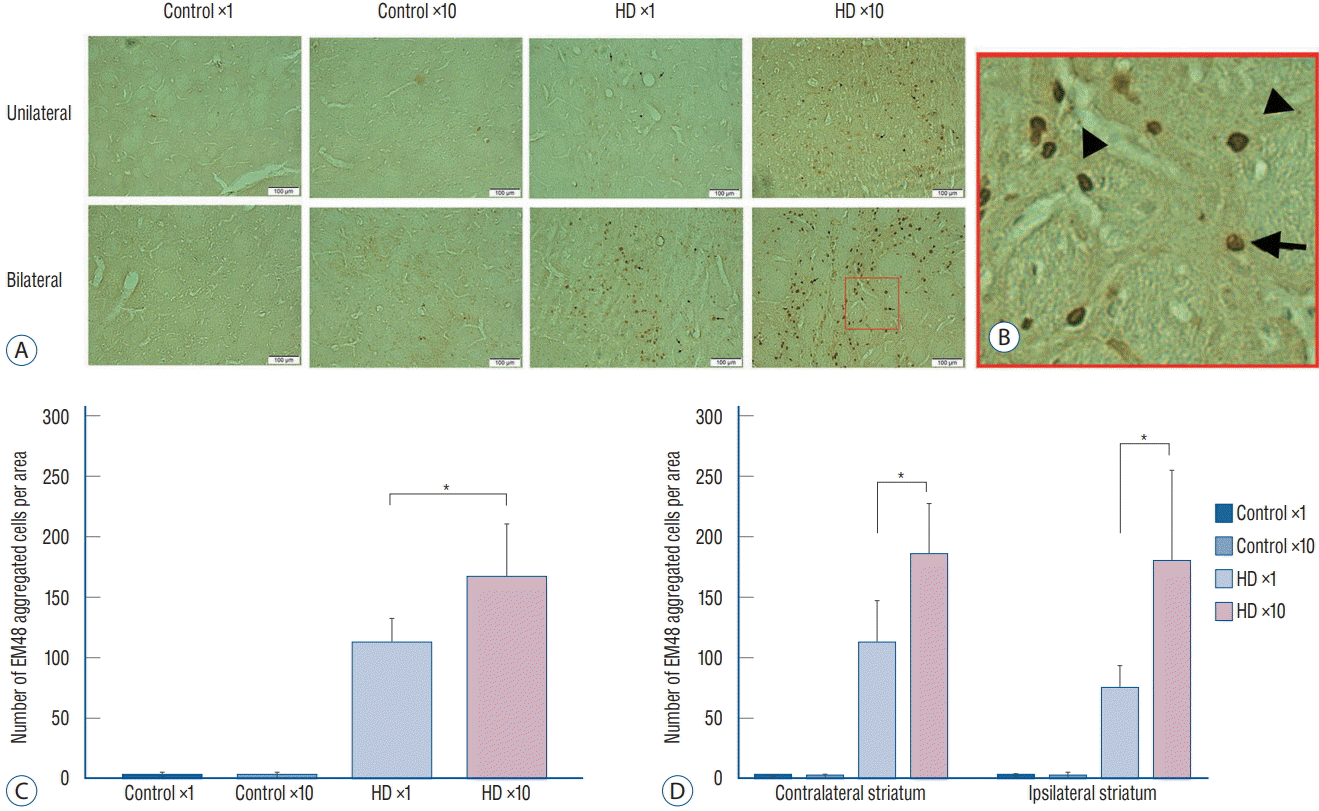
We injected 2 μL of PBS into the contralateral striatum and 2 μL of AAV2-82Q into the ipsilateral striatum of the unilateral ×1 and ×10 subgroups, whereas 2 μL of AAV2-82Q were injected into both sides in the bilateral ×1 and ×10 subgroups. We counted the number of cells expressing EM48 under the microscope at 20× magnification. The mean number of cells counted in both striata of the bilateral injection group was significantly increased in the ×10 subgroup compared with the ×1 subgroup (right hemisphere : 74±19.78 vs. 180±76.11, p=0.035; left hemisphere : 184±43.58 vs. 111±36.48, p=0.010). In the unilateral group, the number of EM48-positive cells in the right striatum of the ×10 subgroup was significantly higher than in the ×1 subgroup (165.83±45.15 vs. 110.5±21.69, p=0.029) (Fig. 4). Few cells were labeled with the EM48 antibody in the control subgroup and the contralateral striatum of the unilateral group.
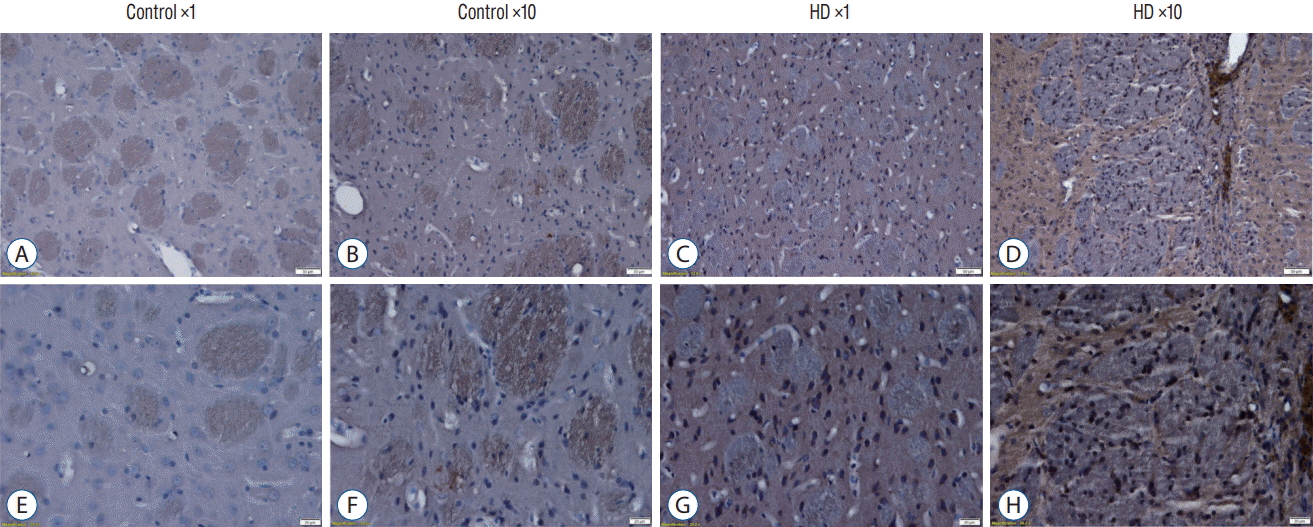
We also detect the expression of huntingtin protein by using anti-huntingtin protein antibody (clone 1HU-4C8, mAb2166) (Fig. 5). The mAb2166 positive cells were found with higher expression in the AAV2-82Q ×10 injected subgroup (Fig. 5D and H) than in the ×1 subgroup (Fig. 5C and G). The mAb2166 positive cells was scarcely found in the control subgroups (Fig. 5A, B, E, and F).
The effect of AAV2 infection into microglia, astrocytes, oligodendrocytes, and neurons was shown in Fig. 6. The brain sections showed the activation of microglia (Iba-1 positive) and astrocytes (GFAP positive) in AAV-82Q-injected rats, and more positive cells was observed in the injected groups compared to the control groups. Also, more positive cells were detected in the ×10 subgroup than in the ×1 subgroup. Higher expression of an oligodendrocyte marker (Olig2) and an apoptosis marker (Caspase3) were also detected in the AAV2- 82Q ×10 group than in the control groups and ×1 group. For detection of neuronal dysfunction, the neuronal cell marker (NeuN) was stained that the expressions were slight reduced at HD groups than control groups whereas there were no significant differences between control and mutant groups.
DISCUSSION
In this study, we developed a new Huntington rat model by directly injecting the caudate putamen with two different viral titers of AAV2-82Q (2×1012 or 2×1013 GC/mL) to determine the optimum viral vector titer to induce an HD model. We performed locomotor behavioral testing, magnetic resonance imaging scanning, and a neuropathological study to verify that the model was appropriately established.
First, we identified behavioral changes similar to a patient with HD in the affected limb of the rats. The site in the caudate putamen where we injected AAV2-82Q is the brain structure related to locomotion, learning ability, and anxiety disorders [1,21]. Previous studies using Parkinson rat models have shown decreased motor function in the stepping test due to damage to the putamen [19]. We also injected AAV-82Q into the caudate putamen and conducted a stepping test to evaluate motor function. The number of steps in the unilateral and bilateral groups was significantly decreased compared to the control group, and the decrease was exacerbated in rats injected with a higher viral titer. In addition, we performed a cylinder test to assess locomotor dysfunction. The unilaterally injected rats used the ipsilateral right paw more frequently because the right putamen was damaged by AAV2-82Q. However, in the bilateral injection group, dominant use of the right or left paw was not observed, because the bilateral group was injected with AAV2 in both caudate putamens and exhibited did not exhibit more destruction on one side than on the opposite side. Thus, each rat randomly used their preferred paw depending on the dominantly destroyed caudate putamen. These results confirmed impairments in movement and locomotion due to caudate putamen damage.
Second, we produced brain tissue sections and performed immunohistochemistry to histologically verify the presence of mutant HTT, which is the hallmark of HD, and the effect of the AAV2-82Q injection on neurons, microglia, oligodendrocytes and astrocytes. EM48, a fusion protein antibody directed against the N-terminal region of HTT, was densely aggregated in the AAV2-82Q injection site of the unilateral and bilateral injection groups, whereas little or no aggregation of EM48 was observed in the PBS injection site of the control group. The amount of EM48 aggregation significantly increased with the AAV2-82Q concentration. Our histology results also showed microglia and astrocyte activation in AAV2-82Q-injected rats. This result is similar to the in vivo mouse HD model obtained by stereotaxic infection of AAVDJ containing N171-82Q, which showed that the viral vector infects neurons, microglia, and astrocytes, but not oligodendrocytes [12].
Based on these results, we confirm the possibility of a new Huntington rat model produced by direct infection of AAV2-82Q into the striatum. We believe that these animal models overcome the shortcomings of drug induction or other viral vector models.
The drugs used to construct Huntington animal models, such as kainite, quinolinic acid, and 3-nitroproprionic acid, inhibit the mitochondrial energy metabolic enzyme succinate dehydrogenase, which causes cell death by inducing excitotoxicity and subsequent neuronal damage. Consequently, these drugs result in animal models with motor dysfunction and brain pathology similar to HD [8,27]. Although the mechanism underlying cell death in the drug-induced models may include a related molecular striatal degeneration pathway, the genetic nature of HD has always called into question the relevance of these neurotoxic lesion models [20]. Nongenetic models typically induce cell death either by excitotoxic mechanisms or disrupting the mitochondrial machinery [22]. A clear association between the mechanisms of action of excitotoxins and mitochondrial inhibitors and the genetic cause of HD has not been identified [22].
Several transgenic mouse models have been introduced to overcome this drawback of neurotoxic lesion models [31,33,34]. These models express genomic DNA or cDNAs encoding mutant HTT under the control of endogenous promoters and display many of the typical pathological features of early HD [9,16,24,28,30,35,36]. However, they do not replicate the typical pattern of neurodegeneration, such as the loss of spiny medium neurons, even in transgenic mice expressing full-length HTT [9,23]. Moreover, fatal adverse effects attributed to overexpression of HTT and premature death that is not related to the striatal lesions have been reported [15,23,25].
Recently, animal models using lentiviral vectors encoding mutant HTT have been developed. Lentiviral vectors enable the insertion of target genes into undifferentiated cells and have large vector capacities. However, these vectors have some disadvantages, such as unpredictable loci of viral progeny insertion and the induction of host immune responses [2,6,13,17]. Because AAV induces a weak immune response compared to lentiviruses, it can be more safely introduced into primates [20]. Therefore, the use of AAV can be expanded to other primate models, such as a macaque model, which is essential for studies of human diseases. Additionally, this virus has been inserted into both differentiated and undifferentiated cells and exists as an episomal vector, which allows long-term gene expression [5,18]. Jang et al. [12] have been generated an HD mice model using AAV-DJ-N171-82Q mHTT. AAV-DJ and AAV2 have very strong tropism for neurons and fast expression. However, the expression of AAV-DJ can vary a lot depending on the viral titer and purity, while the expression of AAV2 is clearly known.
Although transgenic rodent models and knock-in mice have been produced, these models are not representative of the genetics of the disease in humans, as patients usually have only one normal copy of the HTT gene and one mutated copy. Moreover, additional pathology not associated with this disease has been observed in these models [22]. The models also have limitations because they are time-consuming and are unlikely to be cost-effective in large mammalian or non-human primate models.
Because the creation of transgenic non-human primates is both time-consuming and costly, the proposed model is particularly attractive for examining complex cognitive and motor behaviors that can best be studied in non-human primates [22]. A human gene delivery method in an HD model is relatively cost-effective and may not introduce additional pathology, as opposed to transgenic methods; thus, this approach can be widely applied to non-human primates for Huntington research. The proposed method is expected to produce stable rodent or non-human primate Huntington models in the same manner.
In the recent study, we provided two different data sets including behavior tests and histological properties of HD rat models that induced by infecting two doses of AAV2-H171-82Q. It can be used as a basis for translating to large animal HD model.
This study has some limitations. Patients with HD display various numbers of CAG repeats and therefore the AAV-82Q 2×1013 GC/mL infection rat model may not represent a universal Huntington model. Moreover, we did not evaluate specific neuronal markers, such as medium spiny neuron (MSN 1) and gamma-aminobutyric acid (GABA 1), in the striatum and cerebral cortex.
CONCLUSION
The injection of 2×1013 GC/mL of AAV2-82Q into the rat striatum generates the optimum Huntington rat model rather than the infection of 2×1012 GC/mL of AAV2-82Q, and thus this model overcomes the shortcomings of other Huntington’s animal models. The delivery of 2×1013 GC/mL of human the AAV2-82 gene resulted in Huntington behaviors and optimum HTT protein expression in vivo. This study would propel to study cell or gene therapy, and overcome disadvantages of existing animal modeling in incurable Huntington disease.

 XML Download
XML Download