

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The discovery of the mechanistic target of rapamycin (mTOR) began with the identification of new antimicrobial agents in soil samples from Rapa Nui (also known as Easter Island) [93]. This new antimicrobial agent, called rapamycin (clinically called sirolimus), was found to exhibit immunosuppressive and anti-cancer potential and to be of use as an antiepileptic drug [43,47,144]. For two decades, endeavors to define the function of mTOR revealed that mTOR coordinates environmental signals and metabolic activity by forming two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [130]. mTOR was first found in brain lysates, suggesting the importance of the brain-specific function of mTOR [125]. Indeed, the mTOR pathway has been found to regulate a variety of functions in the brain from brain development to degeneration [85].
Human genetic studies of epileptic patients and epilepsy models of aberrant activation of the mTOR pathway have demonstrated the importance of activating mutations in the mTOR pathway in neurodevelopmental disorders with epilepsy [66]. With the discovery of mutations activating the mTOR pathway as genetic causes of medically intractable epilepsy [4], mTOR inhibitors, such as sirolimus and everolimus, have emerged as being of potential medical use in treating intractable epilepsy patients [33]. However, with the limited efficacy and significant drawbacks of clinically available mTOR inhibitors, new drugs that are more effective and tolerable based on the understanding of pathogenic mechanisms are required. Key discoveries in research on the role of the mTOR pathway in epilepsy are summarized in Supplementary Box 1.
THE MTOR PATHWAY
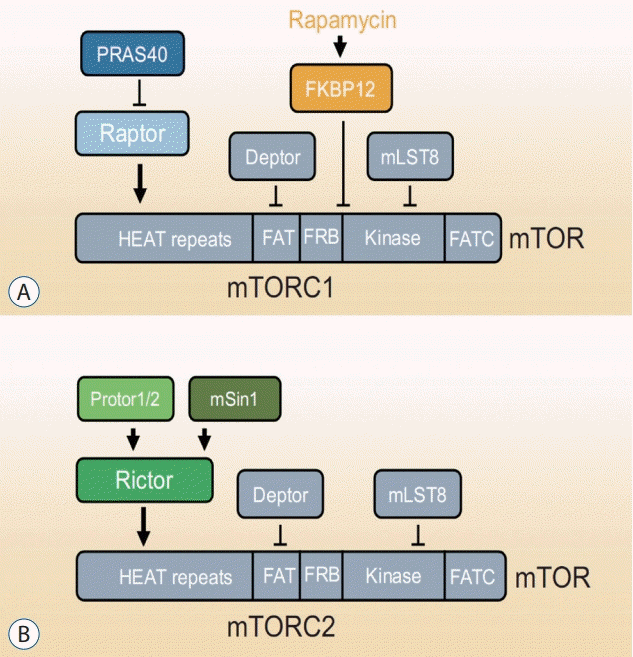
mTOR is a serine/threonine protein kinase that forms the core subunit of two functionally distinct protein complexes : mTORC1 and mTORC2 [130] (Fig. 1). mTORC1 comprises three core components : mTOR, Raptor (regulatory protein associated with mTOR), and mLST8 (mammalian lethal with Sec13 protein 8, also known as GßL) [130]. In response to environmental signals, mTORC1 acts to control the balance between anabolism and catabolism [130]. mTORC2 controls proliferation, survival, and cytoskeleton organization [130]. In a nutrient-rich environment, cells convert energy sources into macromolecules, such as proteins, lipids, and nucleotides. In nutrientstarved environments, however, cells downregulate the production of macromolecules and rely on catabolic pathways, such as autophagy, for energy.
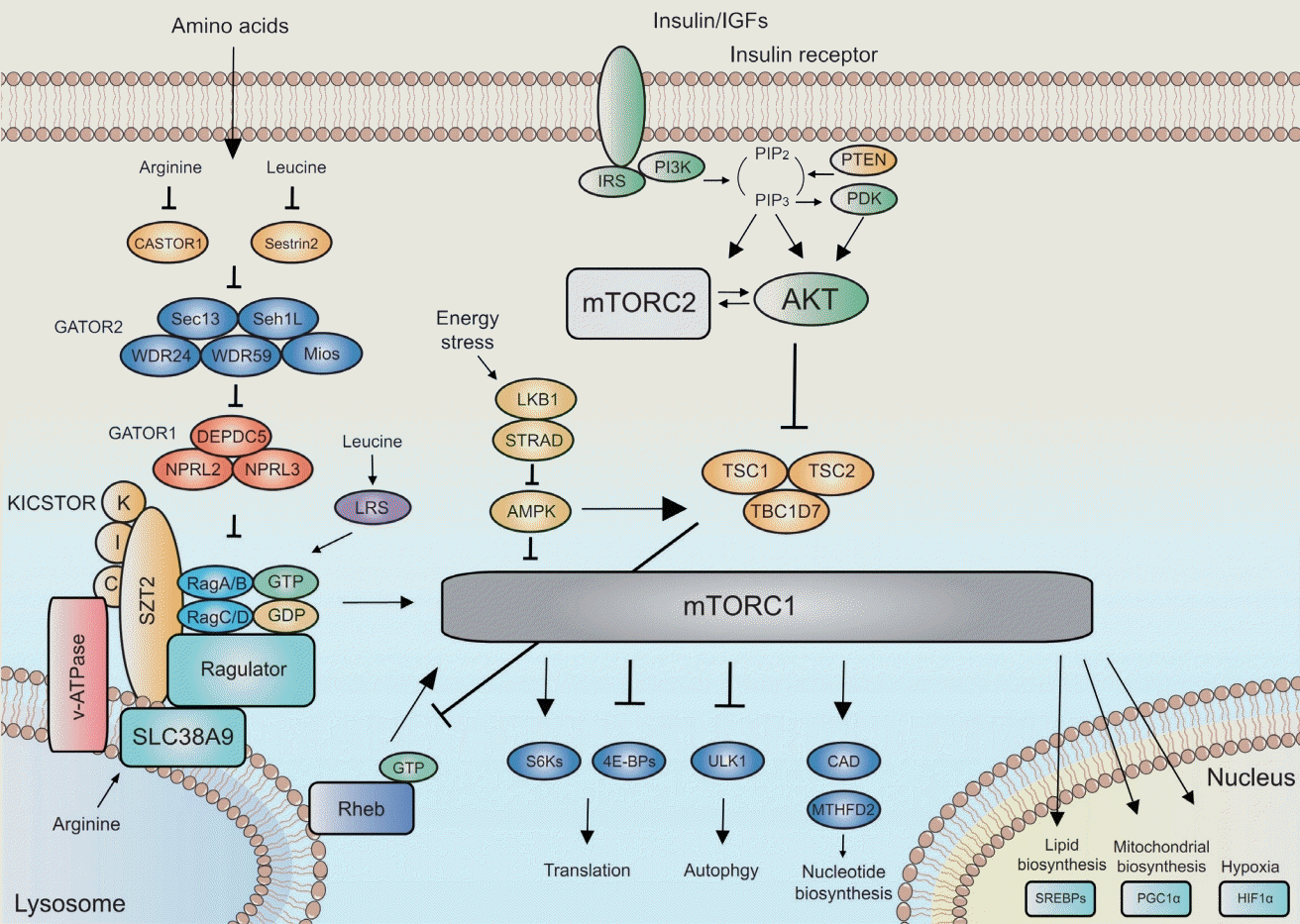
The mTOR pathway receives various inputs from upstream signaling pathways in response to growth factors, amino acids, energy, oxygen, and stress [130], and the upstream pathways of mTOR can be divided as energy-sensing PI3K-PTEN-AKT-TSC and amino acid-sensing GATOR2-GATOR1-Rag GTPase pathways (Fig. 2) : phosphatidylinositol-3-kinase (PI3K) is critical to integrating insulin signaling for growth and survival [130]. PTEN antagonizes the action of PI3K. Akt is activated by PI3K and is a positive regulator of mTORC1 via inhibition of Tuberous Sclerosis Complex (TSC). TSC is a heterotrimeric complex comprising TSC1, TSC2, and TBC1D7 [38]. TSC inhibits mTORC1 by acting as a GTPase activating protein for Ras homolog enriched in brain (Rheb) [67]. Rheb is a small GTPase that activates mTORC1 by directly binding to mTORC1 on the surface of lysosomes [88]. Meanwhile, Rag GTPase, a component of the amino acid sensing pathway [127], activates mTORC1 by promoting translocation of mTORC1 to the lysosomal surface. Upstream regulators of Rag GTPase in amino acid signaling are the GATOR1 and GATOR2 complexes [10]. The GATOR1 complex, consisting of DEPDC5, Nprl2, and Nprl3, inhibits the mTORC1 pathway by acting as a guanine exchange factor for Rag GTPase. The GATOR2 complex, consisting of Mios, WDR24, WDR59, Seh1L, and Sec13, is a positive regulator of the mTORC1 pathway by inhibiting GATOR1. KICSTOR, which is composed of four proteins, KPTN, ITFG2, C12orf66, and SZT2, recruits GATOR1 to the lysosome to inhibit Rag GTPase [150]. Leucyl-tRNA synthetase, which is another amino acid sensor, functions as a GTPase activating protein for Rag GTPase [57]. mTORC1 senses amino acids in an intra-lysosome fashion. Lysosomal amino acid regulates Rag GTPase via v-ATPase, which increases the guanine exchange factor activity of Ragulator towards Rag GTPase [159]. SLC38A9 is a sensor of lysosomal arginine and activates mTORC1 [69]. Additional novel mTOR upstream regulators, including a methionine sensor, have recently been discovered [1,55].
For macromolecule metabolism, mTORC1 regulates translation through inhibitory eukaryotic initiation factor 4E (eIF4E)-binding protein 1/2/3 (4E-BPs) and the S6 kinases (S6Ks) [21,50]. Translational control occurs predominantly at the initiation step, which commences with the binding of the eukaryotic translation initiation factor 4F (eIF4F) complex to the 5’cap [52,135]. As the limiting component of the eIF4F complex, eIF4E is considered to be a critical determinant in translation of mRNA [37]. Facilitating eIF4F formation and the progression of translation, mTORC1 phosphorylates (inactivates) the 4E-BPs, leading to their dissociation from eIF4E [51,60]. The S6Ks activate the eukaryotic translation initiation factor 4B (eIF4B), which is an activator of the eukaryotic translation initiation factor 4A, leading to an increase in the helicase activity of eIF4A and the initiation of translation [39,61].
Stimulating lipid synthesis, mTORC1 interacts with the sterol responsive element binding proteins transcription factors [116]. For sufficient supply of nucleotides during growth, mTORC1 promotes purine and pyrimidine nucleotide biosynthesis through MTHFD2 and the carbamoyl-phosphate synthetase [17]. Through increased translation of the HIF1α transcription factor that drives the expression of glycolytic enzymes, mTORC1 further promotes growth by changing glucose metabolism from oxidative phosphorylation to glycolysis [130]. The mTORC1 pathway also activates the transcriptional coactivator PGC1α for increased mitochondrial biosynthesis [34]. Meanwhile, mTORC1 inhibits autophagy, which plays an important role in scavenging damaged and harmful cellular structures and sustains energy homeostasis, through ULK1 [118]. Recently, it was also demonstrated that mTORC1 regulates ribosomal protein degradation through NUFIP1 [151].
mTORC2 largely functions as a primary effector of the insulin/PI3K signaling [130] (Fig. 2). Generally, the role of the mTORC2 pathway is thought to overlap with the insulin signaling pathway, considering that phenotypes caused by perturbation of the mTORC2 pathway are very similar to those elicited by perturbation of insulin signaling [105]. Upon phosphorylation by mTORC2 [128], Akt promotes cell survival and proliferation. mTORC2 further regulates cellular proliferation and survival through the AGC (PKA/PKG/PKC) family of protein kinases.
PHYSIOLOGICAL ROLES OF THE MTOR PATHWAY IN THE BRAIN
The mTOR pathway plays important roles in development and aging of the central nervous system (CNS) : the mTORC1 pathway is a core regulator of neural development, cortical architecture, neuronal morphology, circuit formation, synaptic plasticity, and neurodegeneration [85]. Perturbation of the mTOR pathway has been found to disrupt several developmental processes. Defective telencephalic development was reported in mTOR mutant rats, called “flat-top” mutants [59]. The loss of function of tuberous sclerosis complex 2 (Tsc2), which is an upstream inhibitor of mTOR, was found to disrupt neuroepithelial growth [119]. Altogether, these studies demonstrated that the mTOR pathway is important for brain development by regulating neural stem cells. Meanwhile, activation of the mTOR pathway via conditional deletion of Tsc2 was found to lead to the disruption of cortical layering [147]. Additionally, increased activity of the mTOR pathway due to mTOR mutations, loss of Tsc1/2, or Pten, was described as eliciting hypertrophy of soma, fewer dendritic spine, increased axon length, and increased dendritic complexity in cortical neurons [53,77,78,137,138].
Perturbation of the mTOR pathway has also been found to adversely affect neural circuit formation. The mTOR pathway regulates axon length [53] and axon guidance in response to environmental signals [156]. Among neurons, local protein synthesis in synapses distant from the soma is mediated by mTOR and is critical for the formation of the neural circuit. The expression levels of synaptic proteins, such as the Arc and Synapsin, are increased by activation of mTOR [81,117]. Hyperactivation of the mTOR pathway by loss of Pten has been shown to increase glutamatergic and GABAergic signals [148]. In addition to neurons, the mTOR pathway has been found to regulate glial cells during neural circuit formation. Deletion of the core component of the mTORC1 or the mTORC2, Raptor or Rictor, was reported to result in defective myelination and oligodendrocyte maturation [18,146]. Conditional Tsc1 knockout in the astrocyte reportedly disrupted electrical signaling in the mouse brain by abrogating neural circuitry [141]. Interestingly, researchers have discovered crosstalk between neurons and glial cells as evidenced by a loss of myelination upon loss of TSC1 in neurons [96]. In TSC patients, the critical role of the mTOR pathway in neural circuit formation was echoed by reductions in white matter and cortical connectivity [112].
Neuronal synapses are crucial to storing memories. Observations of impaired memory formation and synaptic function in response to rapamycin treatment suggest the important role of the mTOR pathway in memory formation [25]. Indeed, the mTOR pathway has been found to play a crucial role in both long-term potentiation (LTP) and long-term depression (LTD) : activation of the mTOR pathway via Tsc1+/-, Tsc2+/-, or 4E-BP2 knockout (KO), results in a decrease in LTP threshold [8,41,145]. Meanwhile, reduced mTOR pathway activity disrupts LTD through metabotropic glutamate receptors (mGluRs) that disturb local protein synthesis [62,64].
ACTIVATION OF MTOR PATHWAY IN NEURODEVELOPMENTAL DISORDERS WITH EPILEPSY
Neurodevelopmental disorders, such as malformations of cortical development (MCD), Cowden syndrome (CS), PIK-3CA-related overgrowth spectrum (PROS), and hamartoma tumor syndromes, commonly present cortical abnormalities and are highly associated with epilepsy, developmental delay, and autism-spectrum disorders [4,12,70,80,97,106,133]. These neurodevelopmental disorders with epilepsy are a common cause of drug-resistant epilepsy in children requiring surgery for treatment [4,12]. However, about 50% of these individuals continue to experience seizures after surgical resection of the epileptic focus [94]. In addition to clinical intractability, refractory epileptic seizures also pose tremendous socioeconomic burden, based on poor quality of life, to patients and their caregivers [28].
Aberrant activation of the mTOR pathway has been identified in brain lesions from patients with neurodevelopmental disorders with epilepsy, especially in MCD [15,86,100]. With these results, researchers have suspected genetic mutations as culprits of the observed mTOR pathway activation in neurodevelopmental disorders [32]. Indeed, a number of neurodevelopmental disorders with epilepsy has been shown to be caused by germline or somatic mutations in the mTOR pathway : these mutations can be categorized as those affecting the energy-sensing PI3K-PTEN-AKT-TSC pathway and those affecting the amino acid-sensing GATOR2-GATOR1-Rag GTPase pathway (Table 1).
MUTATIONS IN THE ENERGY-SENSING PI3K-PTEN-AKT-TSC PATHWAY
Germline mutations in the PIK3CA gene, which activates the mTOR pathway, have been reported in CS [106]. Characterized by hamartomatous overgrowth of tissues, CS is also accompanied by epileptic seizure in a subset of patients [98,113]. There are also other brain and body overgrowth disorders with epilepsy that are caused by post-zygotic mutations in the PIK3CA pathway, which are termed PROS [70]. Additionally, mosaic or brain somatic mutations in PIK3CA have been reported in several other neurodevelopmental disorders with epilepsy, including the megalencephaly (MEG), hemimegalencephaly (HME), megalencephaly-capillary malformation (MCAP), megalencephaly-polydactyly-polymicrogyria-hydrocephalus syndromes, and focal cortical dysplasia (FCD) type IIa/IIb [36,68,79,99,122,132].
Classically, germline PTEN mutation has been reported in hamartoma tumor syndromes, including CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like conditions that share the pathological phenotypes of macrocephaly and megalencephaly [97]. Epileptic seizures have been reported in patients with germline mutation in PTEN [29,30,90].
Brain somatic mutations in AKT3 have primarily been detected in MCD, including the HME and FCD. Brain somatic gain of function mutations in AKT3 were first demonstrated to cause HME [79], followed by identification of somatic AKT3 mutations in FCD [68]. Germline AKT3 mutations were also reported in MEG [104]. To date, germline AKT3 mutations have been reported in HME, MEG, MEG with polymicrogyria, and MEG with periventricular nodular heterotopia [2].
TSC1 and TSC2 germline mutations have been identified in TSC patients [142]. Brain somatic mutations in TSC1 and TSC2 have also been discovered in FCD type IIb [82]. Homozygous TBC1D7 loss of function mutation have been identified in MEG patients [3,22]. More recently, brain somatic mutation in RHEB were identified in an HME patient [126].
STE20-related kinase adaptor α (STRADA) is a negative regulator of mTOR, by activating AMPK [107]. A loss-of-function mutation in STRADA was reported in autosomal recessive disease polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome, also called Pretzel syndrome (PMSE) [111].
Germline or mosaic mutation in MTOR has been described in the Smith-Kingsmore syndrome (SKS) OMIM #616638, which is characterized by epileptic seizure and neurodevelopmental defect [54]. SKS is a rare disease for which only 23 patients have been reported as of 2018 [54,101,102]. Brain somatic mutations in MTOR have also been described in FCD and HME, with varying degrees of mutational burden [91]. All of these mutations have been found to be missense gain-of-function mutations that activate the mTOR pathway.
MUTATIONS IN THE AMINO ACID-SENSING GATOR2-GATOR1-RAG GTPASE PATHWAY
Amino acid-sensing pathway is mediated by the GATOR2-GATOR1-Rag GTPase pathway. Since GATOR1 is a negative regulator of the mTORC1 pathway, researchers proposed that loss-of-function of GATOR1 would lead to neurodevelopmental disorders with epilepsy via hyperactivation of the mTORC1 pathway. Germline mutations in DEPDC5, NPRL2, and NPRL3 have been reported in familial focal epilepsy with variable foci, autosomal dominant nocturnal epilepsy, temporal lobe epilepsy, and familial mesial temporal lobe epilepsy [13,121]. In addition, germline mutations in DEPDC5, NPRL2, and NPRL3 have also been discovered in MCD and epileptic spasms patients [24,131,134]. Additionally, mosaicism and brain somatic mutation in GATOR1 have been reported in MCD [36], pachygyria [26] and familial focal epilepsy with focal cortical dysplasia [14]. Interestingly, it was recently demonstrated that epilepsy patients with focal cortical dysplasia carry a germline mutation in one allele of DEPDC5 and a brain somatic mutation in the other allele, suggesting a two-hit hypothesis as a disease mechanism [14,120]. Autosomal recessive mutations in the KICSTOR complex, including KPTN [9,108], SZT2 [11,143], and the genomic locus that contains C12orf66 [95], have been identified in neurodevelopmental disorders with epilepsy. Until now, mutations in GATOR2 have not been identified in epilepsy patients.
EPILEPSY ANIMAL MODELS OF HYPERACTIVATION OF THE MTOR PATHWAY
For extensive research into epilepsy, genetic animal models of mutations activating the mTOR pathway in epilepsy patients have been developed (Table 2). The first genetic model of epilepsy was the Pten KO mouse reported in 2001 [6,78]. Although Eker rats with germline mutations in the Tsc2 gene were the first genetic animal model of mTOR hyperactivation, the model was not suitable for analyzing epileptic seizures due to early phase lethality [155]. In 2002, the first TSC model with epilepsy, which was the astrocyte-specific Tsc1 KO model, was reported [141]; neuron-specific KO of Tsc1 was also found to lead to spontaneous seizure in the mouse model [96]. After these, the Tsc2 GFAP KO mouse model was developed and shown to lead to epilepsy [157]. A mouse model expressing human PI3KCA mutation in developing neural progenitors was generated to recapitulate pathological features of PROS syndrome, including epilepsy [123]. A knock-in mouse model with gain-of-function mutation in Akt3 has also been found to elicit spontaneous seizure [140].
Recently, epilepsy models of brain somatic mutations, which recapitulate focal malformations of cortical development (FMCD), have been reported. A mouse model of FMCD was first generated by in utero electroporation, thereby providing a small portion of neurons to express known mTOR mutations in human FMCD patients [7,83,110]; these models developed spontaneous seizures. Additionally, FMCD models of brain somatic mutation in mTOR pathway genes, such as RHEB or TSC, have consistently recapitulated spontaneous seizures [63,82]. Heterozygote GATOR1 gene (Depdc5, Nprl2, and Nprl3) KO animals have not been found to recapitulate epileptic seizures observed in patients with germline mutations in the GATOR1 genes [40,65,72,92]. Interestingly, FMCD mouse model caused by a biallelic two-hit mutation (brain somatic and germline) in Depdc5 was shown to lead to spontaneous seizure [120]. Homozygous KO of Szt2, a component of the KICSTOR complex, has been reported as leading to low seizure threshold in mice [46].
MTOR PATHWAY ACTIVATION IN EPILEPTOGENESIS
Epileptogenesis refers to the process leading to the first spontaneous seizure after pro-epileptogenic insult (e.g., genetic defect, brain injury, status epilepticus, or cancer). Epileptogenesis is a brain-wide circuit rewiring process comprising reorganization of microcircuits to long-range circuits, as well as gliosis, blood-brain barrier damage, inflammation, and neurodegeneration [114].
Since epilepsy is the consequence of electrical changes, electrophysiological characteristics in animal models and epilepsy patients have been analyzed in an attempt to understand the epileptogenic mechanism. Studies have indicated that highfrequency oscillations on electroencephalogram are characteristic of FCD [20]. Organotypic brain slice analysis of resected FCD lesions has revealed an electrographic firing pattern of ictal-like discharges after 4-aminopyridine treatment [5], which decreased with treatment with GABAA receptor antagonist.
In brain slice cultures of hyperactive mTOR signaling, researchers have found both the frequency and amplitude of the miniature excitatory postsynaptic current to be increased in glutamatergic synaptic transmission of the hippocampus [89,138]. In GABAergic neurons, hyperactive mTOR signaling increased evoked synaptic responses in the hippocampus [148]. In a subset of auditory cortical neurons of Pten conditional KO, synaptic inputs from the long-range connections, including the contralateral auditory cortex and thalamus, and local connections were increased [152].
In an epilepsy animal model of brain somatic mutations in the mTOR pathway, mutation-carrying cortical neurons showed an increased capacitance, increased cell size, reduced input resistance indicative of a higher current needed to reach the voltage threshold of the action potential, and increased gain of firing frequency [120]. Also, spontaneous excitatory postsynaptic current frequency was decreased in the mTOR pathway mutation-carrying cortical neurons [84,120].
There have been tremendous advances in the identification of the contribution of mTOR downstream functions to neuronal hypertrophy, dendritic branching, axon length, and neuronal migration. Although numerous epilepsy animal models have been utilized to unravel the path to epileptogenesis, the downstream mechanism of mTOR signaling that accounts for epileptogenesis remains unclear. The complexity of the downstream output of the mTOR pathway has hampered attempts to understand the epileptogenic mechanism. Using genetic manipulation techniques, normalizing major downstream outputs of hyperactive mTOR signaling, including translation via overexpressing constitutively active 4E-BP or knockdown of S6Ks [84] and autophagy via knockdown of OFD1 [110], have failed to prevent or even reduce epileptogenesis. Further studies defining the downstream pathway of mTOR in epileptogenesis will be necessary.
INHIBITION OF THE MTOR PATHWAY IN TREATMENT OF EPILEPTIC DISORDERS
Researchers have hypothesized that epilepsy resulting from mTOR pathway activation could be treated by mTOR inhibition, and mTOR inhibition therapies have been evaluated in several clinical trials for epilepsy (Table 3). Rapalogs, including sirolimus and everolimus, have been reported as promising new anti-epileptics because they can penetrate the blood brain-barrier [71]. Rapalogs have been extensively tested and have gained the US Food and Drug Administration (FDA) approval as anti-epileptics in TSC [27,47,73].
The first study to show the beneficial effect of the rapalogs on epilepsy did so in a mouse model of TSC [158]. In this study, early treatment with sirolimus prevented the development of epilepsy. The anti-epileptic effect of rapalogs has also been demonstrated in other genetic models of epilepsy with mTOR pathway hyperactivation, including Pten [87] KO and Strada KO [111]. Recently, intractable epilepsy mouse models with brain somatic mutation in the mTOR pathway were found to be cured by rapalogs [63,82,83]. Interestingly, epileptic seizure was almost completely suppressed by mTOR inhibitors in mouse models of TSC KO or mTOR activating mutation, but only partially suppressed in Pten KO mouse models [87]. These results suggest that an independent mechanism of epileptogenesis distinct from that in TSC KO or mTOR mutation is present in the Pten KO model. In nongenetic seizure models, including the pilocarpine-induced epilepsy model, kainic acid-induced epilepsy model, and absence epilepsy model, rapalogs partially reduced seizures. Citraro et al. [31] reported a representative studies of the anti-epileptic effect of rapalogs in an animal model of intractable epilepsy.
Everolimus have been approved by the FDA for treating subependymal giant astrocytoma, kidney tumors, and partial epilepsy in TSC patients. The first trial to test the efficacy of everolimus to treat epileptic seizure was reported in 2010 for TSC patients [74]. In this study, seizure frequency was reduced in nine of 16 patients with everolimus. Following studies supported the anti-epileptic effects of everolimus or sirolimus (Table 3).
The prospective, randomized, multicenter, placebo-controlled study testing the seizure suppression effect of everolimus on seizure frequency in TSC patients with drug-resistant epilepsy reported positive results [49]. Recently, long-term results of the prospective, open-label, non-randomized study of everolimus for treating epileptic seizure in TSC indicated that reduction in seizure frequency is sustained for up to 4 years [75]. This effect has been confirmed in the long-term follow-up of the EXIST3 trial [35]. Large-scale studies on the anti-epileptic effect of everolimus have described response rates for everolimus and a median seizure frequency reduction in respondents of about 40% [35,75]. In 2017 and 2018, respectively, both the European Medicinal Agency in Europe and the US FDA approved everolimus as an adjunctive therapy in partial-onset seizures in TSC patients aged 2 years and older. However, there are reports of non-responders, seizure aggravation, or an withdrawal after everolimus administration [35,149]. Meanwhile, clinical trials for treating epileptic seizure in FCD type II with everolimus have recently started (clinicaltrials.gov identifier NCT03198949).
The first report of the anti-epileptic effect of sirolimus in TSC patients, which appears to be similar to that of everolimus [23], was in 2009 [103]. In HME patients with brain somatic mutation in mTOR, sirolimus administration reduced seizures [153]. Interestingly, sirolimus has been shown to prevent epilepsy in PMSE patients [111].
Animal and human treatment studies reported that withdrawal of rapalogs provokes seizure recurrence [73]. In electrophysiological analysis of brain slices of resected epileptic foci, everolimus was found to reduce spontaneous excitatory postsynaptic activity, burst discharges, and epileptiform activity in TSC, FCD, and HME cases; effects were subtle in epilepsy patients without mTOR mutations [27].
While rapalogs have been approved for use in various human diseases, concerns have been raised for their long-term use and their adverse effect profile, including potentially serious adverse effects. The adverse effects of rapalogs include immunosuppression, mucositis, hyperlipidemia, hyperglycemia, diabetes-like syndrome, and fatal pneumonitis [42,76,109,139]. In a phase III clinical trial with everolimus, over 90% of 111 patients experienced an adverse effects of any grade [48]. With everolimus treatment for over 1 year, grade 3 or 4 adverse events, which are severe adverse events, were reported in 45% of younger patients and 38% of older patients among 150 patients [35]. We should stress that developmental delay caused by rapalogs [63] should be a concern, considering that the onset of intractable epilepsy with mTOR pathway mutation is usually before adolescence [4]. Additionally, treatment with rapalogs has been found to induce adverse nervous system-specific effects altering sociability [129], learning and memory [16,25], and anxiety [56] in mouse models.
For epilepsy caused by brain somatic mutations, therapeutic regimens must seek to avoid perturbations outside of the CNS, and in this regard, antisense oligonucleotide (ASO) drugs appear to be promising therapeutic options. ASO is a complementary oligonucleotide sequence of sense mRNA sequences that hampers normal gene expression processes, including splicing, transcription, and translation [136]. The characteristics of the bloodbrain barrier prevent ASO from permeating outside of the CNS when administered into the cerebrospinal fluid via intrathecal injection. Moreover, the half-life of ASO drugs is more than 3 months [19]. Recently, ASO drugs have been approved by the FDA for treating various types of neurodegenerative disorders [45]. We suspect that intrathecal injection of ASO drugs targeting mTOR itself, mTOR mutation, or downstream targets of mTOR will prove effective in treating intractable epilepsies while avoiding the adverse effects of rapalogs outside of the CNS.
CONCLUSION
Over the last two decades, extensive research to outline the roles of the mTOR pathway, to identify mutations in the mTOR pathway in human epilepsy patients, and to develop mTOR inhibitors have led to the discovery of novel strategies for diagnosing and treating intractable epilepsy. The overarching goal of this clinical research is attaining seizure-free status with few to no adverse effects in epilepsy patients. While rapalogs have proven to be effective in controlling seizures, complete seizure cessation has not been recorded in most of patients [35,75]. Also, about half of all TSC patients fail to respond to rapalogs. These discrepancies suggest that there might be an unknown biological mechanism at play. Considering about 40% of patients treated with rapalogs experience a severe adverse effect, it will be necessary to develop more potent and tolerable therapies.
While FMCD patients with drug-resistant epilepsy typically undergo surgical resection of the epileptic focus for treating the seizure, up to 50% of patients do not respond to therapy. Based on preclinical and clinical studies of TSC with rapalogs, it may no longer be necessary to perform highly invasive surgical resection for seizure treatment. However, there is a problem in that diagnosing FMCD patients with low-frequency brain somatic mutation in the mTOR pathway requires analyzing DNA from brain tissue after surgical resection. To alleviate this issue, minimally invasive diagnosis through biomarkers in patient cerebrospinal fluid would prove invaluable.
 XML Download
XML Download