

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Rheumatoid arthritis (RA) is an autoimmune disease characterized by articular manifestations, such as persistent inflammation and the progressive degeneration of joints [1]. Approximately 50% of patients with RA show extra-articular symptoms involving organs such as the skin, eyes, heart, kidneys, nerves, gastrointestinal tract, and lungs during the disease course [2]. The lungs of patients with RA often show a wide range of manifestations, such as interstitial lung disease (ILD), infections, and drug toxicity; such pulmonary complications affect the natural course of RA and are consequently associated with increased mortality [1,3]. In particular, approximately 10% of all RA cases are accompanied by severe ILD. The mean survival is 5~8 years, and it is the second-leading cause of death, following cardiovascular disease, in patients with RA [1]. Various patterns of RA-ILD include usual interstitial pneumonia (UIP), nonspecific interstitial pneumonia (NSIP), respiratory bronchiolitis, mixed forms, and unclassifiable interstitial pneumonia, organizing pneumonia, desquamative interstitial pneumonia, lymphoid interstitial pneumonia, and idiopathic pleuroparenchymal fibroelastosis. Among these, two representative histopathological patterns of RA-ILD are UIP and NSIP, and the most common finding is UIP, which has been reported in up to 66% of patients with RA-ILD in previous studies [1]. Lung damage associated with RA-ILD causes irreversible changes and increases mortality in patients with RA; thus, early diagnosis is essential for the successful treatment of patients with RA-ILD [4]. In general, immunosuppressive agents, such as corticosteroids, cyclophosphamide, mycophenolate mofetil, azathioprine, and calcineurin inhibitors, are used for the treatment of RA-ILD. Recently, anti-fibrotic agents, such as pirfenidone and nintedanib, have also been used [1]. However, optimal guidelines for the diagnosis and treatment of RA-ILD have not yet been established [5]. This review aimed to identify risk factors, pathogenesis, experimental methods, early diagnosis modalities, and appropriate therapeutic agents for RA-ILD.
BASIC ASPECTS OF RA-ILD
Pathogenesis of RA-ILD
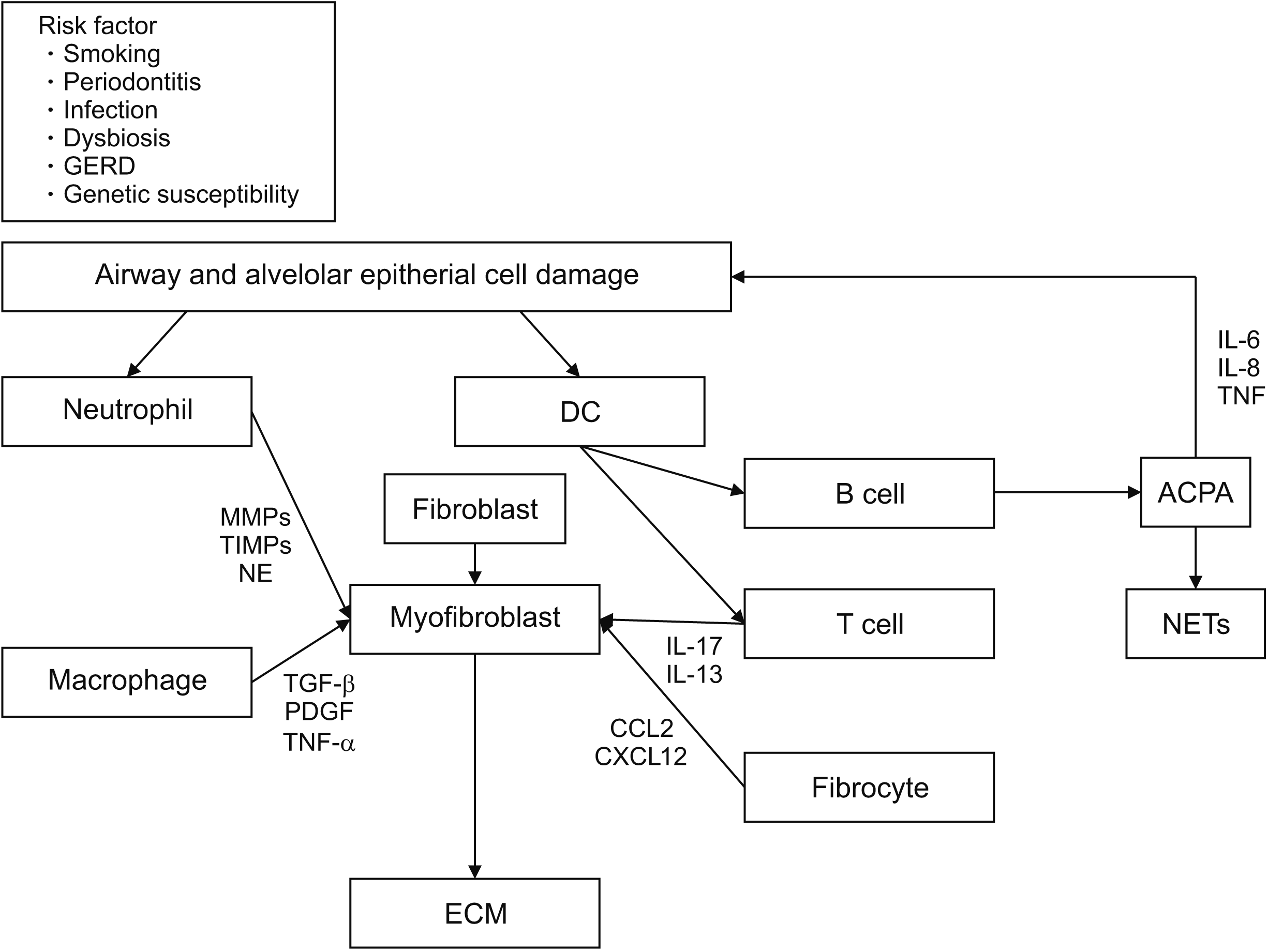
Various genetic, environmental, inflammatory, and immunological factors are involved at each stage of the development and progression of RA-ILD (Figure 1). Several genetic predispositions, such as the expression of human leukocyte antigen (HLA) genes, MUC5B, SFTPC, RTEL1, and TERX, are associated with RA-ILD [6]. Recent studies have also suggested that several HLA variants, including HLA-DRB1*15 and HLA-DRB1*16, are associated with an increased risk of RA-ILD. However, the HLA-DRB1 shared epitope is associated with smoking and reduced risk of RA-ILD [6,7]. MUC5B polymorphism is associated with the abnormal production of surfactant protein C secreted from type 2 alveolar epithelial cells [8]. The mechanism by which excess MUC5B expression results in idiopathic pulmonary fibrosis (IPF) is unknown. However, the abnormal production of surfactant protein C promotes macroautophagy and the secretion of pro-inflammatory and pro-fibrotic mediators from type 2 alveolar epithelial cells, thereby promoting inflammatory responses in the lung parenchyma [6,9]. MUC5B polymorphism acts as a risk factor for IPF and RA-ILD, whereas it is not associated with systemic sclerosis or myositis-associated ILD. Hence, RA-ILD may be associated with IPF more closely than pulmonary fibrosis is associated with other connective tissue diseases [6].
Numerous environmental factors, such as smoking, oxidative stress, infection, and gastroesophageal reflux disease, induce inflammatory stimulation in alveolar epithelial cells [6,10]. Generally, inflammation activates the repair pathway that comprises normal fibroblasts and myofibroblasts, leading to a normal homeostatic response for healing and restoring the lung anatomy [11]. However, prolonged inflammation leads to alveolar and peribronchial wall proliferation, and an abnormal fibrotic repair process leads to the excessive accumulation of extracellular matrix (ECM) components, such as hyaluronan, fibronectin, and interstitial collagens [12]. Various cells are involved in this process. Neutrophils produce neutrophil elastase, matrix metalloproteinases (MMPs), and tissue inhibitors of metalloproteinases (TIMPs). Therefore, they activate transforming growth factor beta (TGF-β) and induce the migration of other inflammatory cells to the lung, thereby promoting pulmonary fibrosis [12,13]. Alveolar macrophages are involved in the pathogenesis of fibrotic lung disease and express pro-fibrotic cytokines, such as TGF-β1 and platelet-derived growth factor (PDGF), chemokines, and proteases, including MMPs. However, alveolar macrophages exert both pro-and anti-fibrotic effects, depending on the local environment and stages of fibrotic diseases [14]. M1 macrophages activate myofibroblast apoptosis and MMP for ECM degradation. Conversely, M2 macrophages secrete TGF-β1 and PDGF to activate fibroblasts, and M2 macrophages secrete TIMPs and inhibit ECM degradation [12]. In case of T-cells, a distorted Th1/Th2 balance plays an important role in lung fibrosis; Th1 and Th22 suppress fibrosis, whereas Th2, Th9, Th17, and Treg aggravate fibrosis [12]. However, there have been conflicting findings on Th17 and Treg cells, and their roles in pulmonary fibrosis remain controversial. Lo et al. showed that Th17 cells exert no direct effect on fibrosis [15]. Findings from other studies have supported the role of interleukin (IL)-17 and Th17 cells by demonstrating that the blockade/neutralization of IL-17A delayed the progression and promoted the resolution of pulmonary fibrosis in different murine fibrosis models [16-19]. Some studies have reported an increase in Tregs in IPF [19], whereas others have demonstrated a reduction in Tregs in the peripheral blood and bronchoalveolar lavage (BAL) fluid in patients with IPF [20]. Fibroblasts are key cells responsible for the synthesis and deposition of ECM, as they provide the initial scaffold for tissue generation in wound healing observed after inflammation [14,21]. Once fibroblasts acquire a pro-fibrotic phenotype with resistance to apoptosis, fibroblasts differentiate into myofibroblasts that sustain the fibrotic process. Both innate and adaptive immune cells are involved in this process [12,14,21]. Fibrocytes produce ECM, cross-linking enzymes, chemokines, growth factors, and MMPs, thereby promoting pulmonary fibrosis [12]. Chemokines, CCL12, and CXCL12 attract circulatory fibrocytes to the site of pulmonary injury [22]. The expression of CXCL13, a B-cell chemoattractant, correlates with the extent of inducible bronchial-associated lymphoid tissue (iBALT) in RA-ILD. However, the role of these chemokines in the development of RA-ILD has not been clearly elucidated [23]. Various other cytokines are also involved in RA-ILD. tumor necrosis factor (TNF)-¥á is a key proinflammatory cytokine in the pathogenesis of interstitial lung involvement. TNF-α stimulates fibroblast proliferation and triggers the expression of PDGF-β, TGF-β, cytokines, and chemokines [23]. IL-1β mediates pro-fibrotic effects through the IL-1R1/MyD88 signaling pathway, and IL-13 is involved in the differentiation of human lung fibroblasts into myofibroblasts through a JNK-dependent pathway. IL-17 interacts with the TGF-β signaling pathway to promote pulmonary fibrosis [12]. Although RA-ILD and IPF pathogeneses have several similarities, further research is necessary to examine the characteristics of RA-ILD pathogenesis.
Roles of anti-citrullinated protein antibody (ACPA) and lungs in the pathogenesis of RA and RA-ILD
ACPA is a collection of autoantibodies with different isotype usage (i.e., IgG, IgA, and IgM) that recognize citrullinated peptides [24]. In the late 1930s, rheumatoid factors were shown to be involved in the onset of RA. Subsequent studies have reported that various antibodies, such as anti-perinuclear, anti-collagen II, anti-hnRNP (RA33), anti-glucose phosphate isomerase, and anti-binding immunoglobulin (BiP) antibodies, may be involved. A study published in 1998 also suggested that auto-antibodies against citrullinated self-proteins and self-peptides, such as homocitrullinated (carbamylated) proteins, may also play a role in the pathogenesis of RA [25]. RA-related antibodies, including ACPA, RF, and carbamylated proteins, are produced several years before the onset of RA joint symptoms and are involved in the development of RA [25]. Constant and repeated damage to the airway mucosa and distal lung structures in the preclinical stages of RA activates the innate immune system and induces inflammatory responses [10]. Such inflammatory responses activate peptidyl arginine deiminase, which leads to increased protein citrullination. Immune tolerance fails in individuals with specific genetic backgrounds, and autoimmune responses induce RA-related ACPA [26]. Persistant inflammation leads to iBALT development, particularly near the airways, perivascular space, and interstitial region. The subsequent activation of local immune cells, such as macrophages and dendritic cells (DCs), and repeated damage to epithelial cells leads to the secretion of other cytokines, such as TGF-β and PDGF. This promotes epithelial-to-mesenchymal transition, remodeling, and fibrosis of the airway and distal lung [26]. Furthermore, ACPA may regulate neutrophil cell death and the secretion of neutrophil extracellular traps, further promoting inflammation and autoimmunity. These autoimmune responses are initially observed only in the lungs; however, these responses may become systemic after additional damage [6].
Numerous studies have shown that the lungs and ACPA are associated with the development of RA. As terms used to designate cases of ACPA-positive fibrotic ILD without clinical symptoms of RA, “lung-limited RA”, “autoimmune-featured ILD”, and “interstitial pneumonia with autoimmune features” have similar meanings [6]. Other evidence supporting the association between the lungs and RA is as follows: smoking may increase the expression of citrullinated proteins in BAL cells, and ectopic lymphoid tissues with B-cells that can bind to citrullinated proteins have also been observed in the lungs of patients with RA. These results suggest that ACPA could be locally expressed in the lungs of patients with RA [27,28]. Moreover, a greater level of ACPA has been observed in BAL fluid than in the blood of untreated patients with ACPA-positive RA [29]. In addition, in patients with risk factors for progression to RA and no joint symptoms, the sputum test showed the local expression of ACPA and RF [30]. Also, in Korean patients with RA, the titer ratio of ACPA was higher in the airway disease group and interstitial lung disease group than in the normal lung group [31]. For this reason, the lung is recognized as a representative organ considered to be related to RA development.
Animal models of RA-ILD
Representative models using immune stimulants, combinations of environmental factors, and genetic engineering methods that have been used in previous studies are summarized in Table 1. Previous reports have shown that pulmonary inflammation accompanying arthritis develops in several autoimmune arthritic mouse models, such as K/BxN arthritis, SKG mice, TNF transgenic (TNF‐Tg) mouse model, and collagen-induced arthritis (CIA) [32]. In K/BxN arthritis, transgenic mice with the KRN T-cell receptor specific for the autoantigen glucose-6–phosphate isomerase (GPI) were crossed with GPI-specific immunoglobulin heavy and light chain knock-in mice. Thus, arthritis develops as a result of the production of pathogenic GPI-specific autoantibodies [33]. Shilling et al. [33] showed that 67% of these mice exhibited lymphocytic infiltration similar to that observed in bronchus-associated lymphoid tissue (BALT) in the lungs, suggesting that systemic autoreactivity promotes ectopic lymphoid tissue development in the lung through the cooperation of autoreactive T and B cells. However, these BALT-like lesions may not be sufficient to promote fibrotic lung disease at a steady state or after an inflammatory challenge [34]. SKG mice have a spontaneous point mutation in the second SH2 domain of ZAP70 relevant to tyrosine-phosphorylated immunoreceptor tyrosine-based activation motifs of TCR‐ζ and CD3 chains [35]. Zymosan is derived from crude yeast cell wall extract, the main constituents of which are β‐glucans [36]. SKG mice were administered a single intraperitoneal injection of zymosan to induce severe arthritis and the advanced onset of lung pathology, strongly resembling NSIP pathology, similar to that observed in human RA‐ILD [36-39]. The TNF‐Tg mouse, overexpressing the human TNF‐α gene, also developed inflammatory erosive arthritis along with a lung abnormality similar to NSIP [32]. CIA leads to polyarthritis and causes chronic and destructive joint damage, which is similar to the symptoms of human RA in many aspects; thus, it is widely used for research on RA [40]. Several reports demonstrated that this model has advantages over other RA mouse models because it shows similar findings to UIP of RA-ILD and helps study the effects of citrullination and ACPA on RA-ILD development [41-43]. The limitation of this method is that the spontaneous resolution of lung lesions occurs after the onset of active and progressive joint inflammation (Figure 2, unpublished data). In contrast, a tendency for the progression and exacerbation of lung diseases is observed in most cases of human RA-ILD. There have been some attempts to use interferon gamma receptor-knockout mice, different species of tree shrews (Tupaia belangeri chinensis), or inhalational organic dust extract combined with CIA, which can be used to develop experimental models showing persistent and progressive pulmonary fibrosis with greater clinical relevance [41,43,44]. Collectively, it is important to establish appropriate experimental animal models to confirm the precise pathogenesis and explore the usefulness of candidate drugs or biomarkers for RA-ILD.
CLINICAL ASPECTS OF RA-ILD
Screening and early diagnosis of RA-ILD
Identifying risk factors is important for RA-ILD screening in patients with RA. In case of ACPA, some reports are related to RA-ILD. Correia et al. [44] reported that a higher titer of ACPA was associated with an increase in the prevalence of ILD (10.3%, 8.82%, 4.92%, and 1.90% in the high, moderate, low, and negative titer groups, respectively, p<0.001) [45]. In addition, in a recent meta-analysis, serum ACPA positivity was strongly associated with RA-associated pulmonary diseases, particularly RA-ILD and IPF. This association was also observed in Korean RA patient subgroups. Other factors related to RA-ILD include older age, male sex, cigarette smoking, RF, ACPA, erythrocyte sedimentation rate, prednisolone use, and disease activity [31,46-49].
The main clinical symptoms of RA-ILD are exertional dyspnea, chronic dry cough, fatigue, and general weakness [49]. However, these symptoms may be overlooked. Therefore, patients with RA must be assessed carefully by checking the accompanying symptoms and RA-associated pulmonary diseases. In clinical practice, various tests, such as pulmonary function testing (PFT) and high-resolution computed tomography (HRCT), are conducted to diagnose and evaluate RA-associated pulmonary diseases. In particular, advances in imaging examinations play an important role in RA-ILD diagnosis [50]. In the past, the prevalence of RA-ILD based on clinical examinations and chest X-rays was 1%~5% [51,52]. The diagnosis rate of RA-ILD is increasing with the development of imaging examinations, such as HRCT. In a recent study that conducted chest PA, PFT, and HRCT in 150 patients with RA, 28 (19%) patients showed signs of fibrosing alveolitis on HRCT [53]. However, even though imaging examinations, such as HRCT, are helpful in the diagnosis of RA-ILD, these examinations are not recommended for the initial screening or routine assessment of patients without respiratory symptoms. Therefore, to diagnose RA-ILD, the respiratory symptoms must be closely monitored, and in patients who have been diagnosed with RA-ILD with aggravated respiratory symptoms, it is necessary to differentiate between infection, drug toxicity, or worsening of existing concomitant diseases [50]. Therefore, the screening and diagnosis of RA-ILD as well as continuous observation of the clinical course and adequate treatment are important. PFT is also important for RA-ILD diagnosis, and the diffusing capacity of the lungs for carbon monoxide (DLCO) is known as the most sensitive test for screening RA-ILD [50]. However, in one study, a decrease in DLCO was observed in 80% of patients with RA, whereas only 5%~15% of patients showed findings of purely restrictive defects on spirometry. Therefore, PFT is not considered cost-effective and is not recommended for routine assessment in patients with RA [6,50]. A recent study suggested a diagnostic algorithm for evaluating RA-ILD. The authors recommended checking the patient history to determine whether dyspnea on exertion is accompanied with climbing stairs or uphill. Thereafter, pulmonary crackles and digital clubbing should be checked through lung auscultation and physical examination, and if these findings are confirmed, DLCO should be measured. HRCT is recommended if DLCO is less than 70%. However, if there are no accompanying respiratory symptoms in patients with RA, the annual screening of symptoms is recommended [50]. Another recommended screening method is plain chest radiography at RA diagnosis, along with additional examinations. However, plain chest radiography is disadvantageous because it has low sensitivity and is limited to the detection of early ILD or subclinical airway disease [6,54]. In recent times, studies have attempted to diagnose ILD using ultrasound and reported that findings of multiple B lines, irregularities of the pleural line, or pleural nodules on ultrasound might suggest ILD [55]. In another study, Manfredi et al. [56] evaluated RA-ILD using a Velcro sound detector. The lung sounds of 137 patients who underwent HRCT were recorded and analyzed using a commercial electronic stethoscope. The accuracy, sensitivity, and specificity of the diagnostic rate were 83.9%, 93.2%, and 76.9%, respectively [56], Other studies have attempted to identify serological biomarkers for screening of RA-ILD. Biomarkers, such as serum ACPAS, MMP7, and CXCL10, and some heat shock proteins have been identified. In the KORAIL Cohort Baseline Data study, not only MMP7, but also SP-D and KL-6 were found to be useful for evaluating the functional and anatomical status of lung involvement in patients with RA-ILD [57]. However, these biomarkers are currently not used in clinical practice [58,59]. Therefore, to screen and diagnose RA-ILD, respiratory symptoms must be evaluated, followed by lung auscultation and plain chest radiography at the time of diagnosis. Additional tests, such as PFTs and HRCT, may need to be performed depending on respiratory symptoms for the clinical diagnosis of RA-ILD. Furthermore, continuous studies on establishing serological biomarkers would further contribute to the screening and early diagnosis of RA-ILD. Based on the reported evidence, the most useful tests for the diagnosis of RA-ILD are HRCT and PFT. These tests should be conducted actively to diagnose RA-ILD.
Treatment of RA-ILD
Disease-modifying anti-rheumatic drugs (DMARDs) that are used to treat RA can be classified into conventional synthetic DMARDs (csDMARDs) and biologic DMARDs (bDMARDs), which have been used increasingly in recent times. Methotrexate (MTX) is a representative csDMARD, and bDMARDs include anti-TNF agents, IL-6 receptor inhibitors (tocilizumab), T cell costimulatory inhibitors (abatacept), and anti-CD20 antibody (rituximab). However, there is controversy regarding the efficacy and safety of these drugs in RA-ILD treatment. Additionally, anti-fibrotic agents, such as nintedanib and pirfenidone, have recently been used for the treatment of RA-ILD. Therefore, first, we will consider DMARDs that can be used safely, following which we will consider anti-fibrotic agents.
MTX, considered the standard treatment agent for RA for long, is not administered to patients with RA-ILD owing to potential pulmonary toxicity and MTX-related hypersensitivity pneumonitis [60-62]. However, unlike findings from initial studies, recent results suggest that DMARDs, such as MTX and LEF, may be used when ILD is accompanied, and these drugs slow the development and progression of ILD [63-65]. In a recent meta-analysis of data from patients administered MTX, there was no increase in non-infectious respiratory adverse events caused by MTX administration [66]. A study on the association between MTX and RA-ILD in two independent British cohorts also reported that MTX was not associated with RA-ILD and MTX delayed RA-ILD development [48]. Additionally, a recent review article, in which 29 articles were analyzed, reported that MTX slowed the progression of ILD and improved the prognosis of RA-ILD [67]. As such, accumulating evidence suggests that cDMARDs, including MTX, may be used to treat patients with RA-ILD. Therefore, unlike in the past, MTX can be used for the treatment of patients with RA-ILD. However, even though various findings have been reported in recent times, further studies must be conducted to fully assess the effects of MTX on the progression of ILD [60].
Rituximab is one of the most important biologics for patients with RA-ILD [61]. Based on the results of various studies in which rituximab was used for the treatment of RA-ILD, it is considered safe for treating patients with RA-ILD. In a study of 43 patients with RA-ILD included in the British Society for Rheumatology Biologics Register for RA, who received rituximab and TNF inhibitors as the first biologic, the mortality rate was lower in the rituximab group [68]. However, in this study, the severity and subtype of ILD could not be assessed. In another observational study of 56 patients with RA-ILD treated with rituximab, 16% showed improvement, whereas 52% showed stable findings. A history of ILD progression was suggested in that study as a progression factor for ILD [69]. In addition, a recent study on 68 patients included in the pneumology Rheumatology Autoimmune diseases registry reported that the frequency of respiratory impairment was lower in the rituximab group than in other groups [70]. Additionally, an EvER-ILD using rituximab with MMF and RITUX-IP study using rituximab are currently underway [60]. Abatacept is considered to be relatively safe for treating RA-ILD. In a retrospective study of 44 abatacept-treated patients with RA-ILD in Italy, 81.4% and 86.1%~91.7% of patients showed stable or improved results on HRCT and PFT, respectively [71]. In a multicenter study of 63 RA-ILD patients treated with abatacept, 11 of 22 patients (50%) who were followed up and underwent HRCT showed stable results, and eight (36.4%) patients showed improved results [72]. A multicenter study in Spain reported that non-TNF inhibitor biologics, including abatacept, slowed the progression of ILD in 69 patients [73]. APRIL, a large-scale study of abatacept, is underway [60]. Tocilizumab may also be used in the treatment of RA-ILD. In a retrospective study of 28 patients with RA-ILD treated with tocilizumab, 56% and 89.3% patients showed stable results for PFTs and HRCT, respectively [74]. However, additional randomized controlled trials (RCTs) will be necessary in future.
Nintedanib was found to be effective for treating progressive fibrosing ILD, including RA-ILD, in a double-blinded RCT [61]. The INBUILD trial was recently conducted on 663 RA patients with 89 ILD patients. In this study, nintedanib slowed the rate of decline in forced vital capacity in patients with RA-ILD, but the sub-analysis of RA-ILD did not show any effects. Based on the results of this study, nintedanib was approved for the treatment of chronic fibrosing progressive ILD, including RA [60,75]. In addition, pirfenidone, used as an antifibrotic agent in IPF, was found to be effective against the UIP pattern. Clinical trials (TRAIL trial, a phase 2 double-blind RCT) for evaluating its safety and efficacy are currently underway [76].
CONCLUSION
RA-ILD is a common extra-articular symptom observed in patients with RA that may develop before the diagnosis of RA or at any time point during the disease. Therefore, it is necessary to check whether RA-ILD is present in patients with RA at RA diagnosis. If there are respiratory symptoms, it is necessary to properly conduct PFT and imaging tests, including HRCT, for early diagnosis. Recent findings suggest that csDMARDs, including MTX, may be used to treat RA in patients with RA-ILD without major restrictions; however, further studies must assess the safety and effects of DMARDs. Based on current findings, bDMARDs, such as rituximab, abatacept, and tocilizumab, are considered relatively safe medications for treating patients with RA-ILD. Additionally, nintedanib, an anti-fibrotic agent, may also be used to treat RA-ILD. RA-ILD is an important complication affecting the prognosis of patients with RA. Therefore, several translational and clinical investigations on RA-ILD must be conducted to improve the prognosis of patients with RA (Table 1). In conclusion, early diagnosis and adequate treatment for RA-ILD are important, and the development of serological biomarkers and appropriate therapeutic drugs for early diagnosis is essential.

 XML Download
XML Download