

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Until the latter part of the 20th century, options for treating patients with heart failure (HF) with reduced ejection fraction (HFrEF) were limited. The handful of drugs that were available provided symptomatic relief but no long-term improvement in outcomes. Insights into the pathophysiology of cardiac dysfunction and pathways leading to the progression of HF have resulted in the development of drugs that can substantially improve the clinical course of patients with HFrEF. As a result, virtually millions of patients around the world have experienced vastly better quality of life and greater longevity. This review provides an overview of the drugs that can modify the clinical course of patients with HFrEF, including description of the pivotal trials that established their usefulness, perspective on how they influence the trajectory of disease, their incorporation in HF management guidelines and practical advice on their use in clinical practice. The review will also cover issues that have arisen regarding implementation and provide a brief look into some of the promising new therapeutic approaches that are in development. Diuretics which offer symptomatic relief but do not modify the trajectory of HFrEF will be not discussed, as excellent reviews on this topic already exist,1) nor will treatments for specific underlying conditions (e.g., amyloidosis) which can cause of HFrEF.
PATHOPHYSIOLOGIC TARGETS FOR DRUGS TO TREAT HEART FAILURE WITH REDUCED EJECTION FRACTION
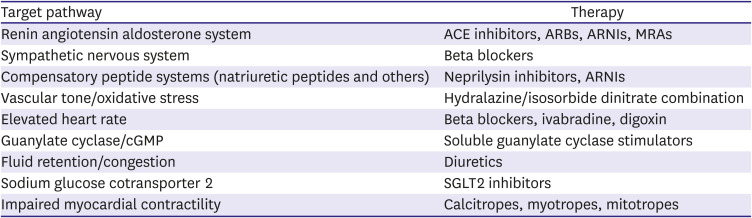
Over the years, a variety of pathways that contribute to the development and progression of HF have been targeted for drug development (Table 1). It is well recognized that HF develops as a consequence of a variety of conditions which injure the myocardium including pressure overload as seen with long-standing hypertension, cardiac cell death caused by a myocardial infarction (MI), viral infection or exposure to toxic substances, and gene mutations that result in structural and functional abnormalities of the heart. Regardless of the inciting cause, injury to the myocardium activates a prototypic response involving the heart as well as other organs around the body. Widespread neurohormonal activation plays a central role in process.2)3)4)5) When sustained over time, it results in circulatory congestion and an increase in pressure and volume load on the heart. Structural changes ensue, including myocardial hypertrophy, dilatation and an increases in interstitial fibrosis, and the left ventricle (LV) assumes a more spherical shape which further compromises contractile function and predispose to secondary valvular incompetence.6) These changes are enhanced by inflammation, abnormalities in ion channel function, dysregulation of calcium flux within cardiac myocytes and impaired mitochondrial energetics.7)8)9)10) Electrical instability predisposes to arrhythmias that contribute to the progressively downhill clinical course. Cardiac dysfunction is further exacerbated by abnormalities that develop in other organs, particularly the vasculature, kidney and skeletal muscle, in parallel with or in response to the hemodynamic abnormalities caused by HF.
Table 1
Pathways for drug therapy for heart failure with reduced ejection fraction
THE CORNERSTONES OF MEDICAL THERAPY FOR PATIENTS WITH HEART FAILURE WITH REDUCED EJECTION FRACTION
Angiotensin converting enzyme inhibitors, angiotensin receptor blockers, and angiotensin receptor-neprilysin inhibitors
Initial attempts at modulating neurohormonal activation involved use of drugs that inhibited angiotensin converting enzyme (ACE), an enzyme that cleaves off the two N-terminal amino acids from the decapeptide angiotensin (Ang) I to form Ang II, the main effector molecule of the renin angiotensin system (RAS). Although other pathways5) including a tissue based one that utilizes chymase for generating Ang II exist,11) ACE is the most important. Upon interacting with its type 1 (AT1) receptor, Ang II activates downstream pathways that are integral to the progression of HFrEF. Early work done by Pfeffer et al.12) demonstrated that ACE inhibition had highly favorable effects on reversing pressure overload induced cardiac remodeling and preventing cardiac dysfunction. Subsequent studies in human patients (summarized in Table 2) demonstrated the effectiveness of blocking RAAS activation in patients with HFrEF.
Table 2
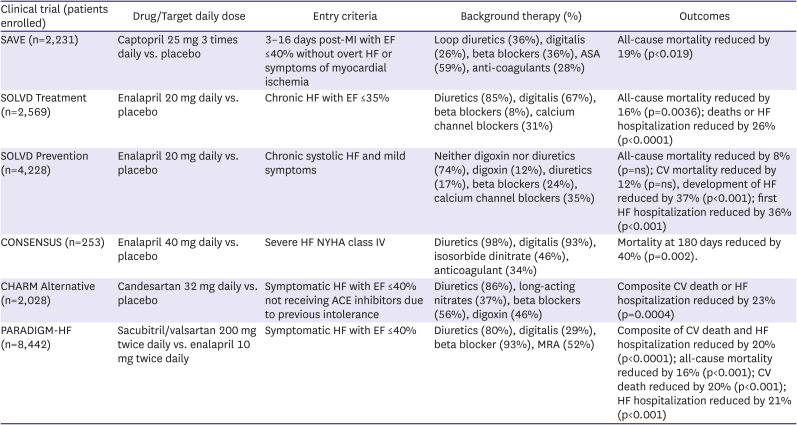
Key clinical trials with ACE inhibitors, ARBs and ARNIs in HF with reduced EF
ACE = angiotensin converting enzyme; ARB = angiotensin receptor blocker; ARNI = angiotensin receptor neprilysin inhibitor; CV = cardiovascular; EF = ejection fraction; HF = heart failure; MI = myocardial infarction; MRA = mineralocorticoid receptor antagonist; ns = not significant; NYHA = New York Heart Association.
The Survival and Ventricular Enlargement (SAVE) trial assessed whether treatment with the ACE inhibitor captopril could improve clinical outcomes in patients following an anterior MI.13) The study randomized 2,231 patients with LV ejection fraction (EF) ≤40% but no overt HF signs and symptoms within 3–16 days after an MI to either captopril or placebo. Captopril treated patients experienced reductions of 20% in all-cause mortality, 37% in risk of developing HF, 22% in risk of HF hospitalization, and 25% in risk of recurrent MI. Subsequent analysis demonstrated the association between progressive post-MI remodeling and clinical events and found an association between attenuation of post-MI LV remodeling by captopril and better outcome.14)15)
The Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) was the first randomized clinical trial to demonstrate that RAS blockade could benefit patients with overt HFrEF.16) The study randomly assigned 253 patients with New York Heart Association (NYHA) class IV symptoms to either enalapril at a target dose of 40 mg daily or placebo. Despite the small sample size and a limited follow-up averaging slightly more than 6 months, there was a significant 27% mortality reduction in treated patients that was due predominantly to a 50% reduction in death due to progressive HF. A significant improvement in NYHA classification was observed with enalapril, together with a reduction in heart size.
The Studies of Left Ventricular Dysfunction (SOLVD) program compared the effects of enalapril at a dose of 20 mg daily vs. placebo on mortality and hospitalization in patients with an EF ≤35%. Patients included in the Treatment Arm of SOLVD had signs and symptoms of HF17) while those included in the Prevention Arm were asymptomatic.18) In SOLVD Treatment, enalapril reduced mortality by 16% with the largest effect being a reduction in death from progressive HF. Combined death and HF hospitalization was reduced by 26%. In SOLVD Prevention, enalapril was associated with insignificant 8% and 12% reductions in all-cause death and cardiovascular (CV) mortality over the 37.4-month follow-up period. There was, however, a highly significant 29% reduction in the development of HF in the enalapril group. Sequential cardiac imaging of patients in the SOLVD trials confirmed the progressive nature of maladaptive LV remodeling and the ability of enalapril to inhibit this process.19)20) Results from the SOLVD registry confirmed the association between increased LV volumes and mass with future events.21) An extensive meta-analysis of placebo controlled trials using a variety of different ACE inhibitors in patients with HF reported a highly significant 23% reduction in total mortality (driven predominantly by a reduction in death due to progressive pump failure) and a 35% reduction in the combined endpoint of mortality or HF hospitalization.22) Benefits were seen in various subgroups including those based on age, sex, NYHA class and etiology of HF.
Angiotensin receptor blockers (ARBs) prevent the interaction between Ang II and the AT1 receptor and they provide an alternative method for RAS inhibition. This strategy was tested in the Candesartan in Heart Failure Assessment of Reduction in Mortality and morbidity (CHARM) Alternative study, a clinical trial in which 2,028 ACE inhibitor intolerant patients were randomized to either candesartan (at a target dose of 32 mg daily) or placebo.23) Over the 33.7-month average follow-up, there was a significant 23% reduction in the composite primary endpoint of CV death or HF hospitalization with the ARB. Candesartan proved to be well tolerated with discontinuation rate similar to that seen with placebo.
Based on these studies, ACE inhibitors were established as a cornerstone of therapy for patients with HFrEF with ARBs considered as an alternative for use in patients already taking an ARB for reasons other than HF or who had limiting side effects with an ACE inhibitor.24) Although both classes of drugs continue to receive a class I recommendation for treating HFrEF in guidelines, the preferred approach for RAS inhibition involves a new class of drugs, the angiotensin receptor-neprilysin inhibitors (ARNIs).25)26)27)
Recognition that neurohormonal activation is widespread in HF focused attention on the potential role of compensatory signaling molecules including adrenomedullin, bradykinin, prostaglandins, apelin and the family of natriuretic peptides, all of which have vasodilatory, diuretic and anti-remodeling effects. In patients with HFrEF, however, these compensatory molecules are insufficient to counter the maladaptive effects of RAS and sympathetic nervous system (SNS) activation. Clearance of the compensatory molecules involves a variety of pathways including metabolism by neprilysin, a membrane bound metalloproteinase that is highly expressed in lung, kidney and elsewhere throughout the body.28) A potential therapeutic strategy is to block the breakdown of the compensatory molecules by inhibiting neprilysin. Initial attempts at this involved omapatrilat, a combined ACE and neutral endopeptidase inhibitor.29) In clinical trials, however, omapatrilat was not superior to enalapril in reducing all-cause mortality and HF hospitalization.30) Further development of omapatrilat was terminated due to an increased risk of angioedema, related to the synergistic effects of dual enzyme inhibition in augmenting bradykinin levels.
An alternative strategy of combining an ARB with a neprilysin inhibitor in the sacubitril/valsartan molecule has proven to be much more successful. In Prospective Comparison of ARNI with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF), a mega-trial of 8,442 patients with symptomatic HF and an EF ≤40%, sacubitril/valsartan at a dose of 97/103 mg twice daily reduced the primary composite endpoint of CV mortality and HF hospitalization by 20% compared to enalapril at 10 mg twice daily.31) This effect was driven equally by reductions in each component and there were significant reductions of 20% in sudden cardiac death, 21% in death due to pump failure and 16% in all-cause mortality.32) Extensive subgroup analysis confirmed the superiority of the ARNI over the ACE inhibitor regardless of age, gender, region, comorbid conditions, etiology of HF, and background therapy. Moreover, the ARNI proved to be more effective than the ACE inhibitor in improving outcomes in patients who were down titrated to a dose below target because of side effects,33) in patients with lower levels of systolic blood pressure,34) in patients with either ischemic or non-ischemic etiologies35) and in older patients.36) While sacubitril/valsartan was associated with a higher likelihood of symptomatic hypotension and non-serious angioedema than the ACE inhibitor, it was also less likely to cause renal impairment, hyperkalemia or cough.
Although sacubitril/valsartan proved superior to enalapril in PARADIGM-HF, questions regarding ARNI use remained. The Comparison of Sacubitril–Valsartan versus Enalapril on Effect on NT-proBNP in Patients Stabilized from an Acute Heart Failure Episode (PIONEER-HF) trial randomized patients shortly after stabilization during a HF hospitalization to sacubitril/valsartan at a target dose of 97/103 mg twice daily or enalapril 10 mg twice daily.37) The primary endpoint was change in N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels over an 8-week period. Although both drugs reduced NT-proBNP levels, the effect was significantly greater with the ARNI which reduced NT-proBNP levels by 29% more than the ACE inhibitor. Clinical events (e.g., composite of death, readmission, heart transplant or need for left ventricular assist device) were 44% lower in the sacubitril/valsartan compared to the enalapril treated patients. Initiation of the ARNI in hospitalized patients also appeared to be safe as the rates of worsening renal function, hyperkalemia, symptomatic hypotension, and angioedema were not greater with sacubitril/valsartan than with enalapril. Unlike PARADIGM-HF which required prior ACE inhibitor or ARB treatment for enrollment, approximately half of the 881 patients randomized in PIONEER-HF were ACEI/ARB naïve. Subgroup analysis demonstrated no difference in effect when patients in whom sacubitril/valsartan was started de novo were compared to those who transitioned from an ACEI/ARB to an ARNI.
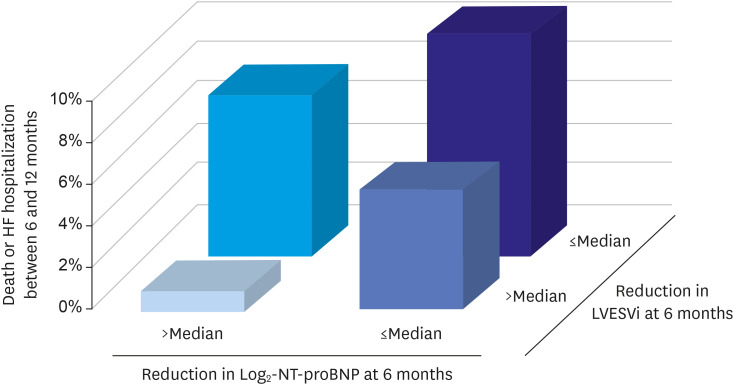
Prospective Study of Biomarkers, Symptom Improvement, and Ventricular Remodeling During Sacubitril/Valsartan Therapy for Heart Failure (PROVE-HF) was designed to assess the effects of sacubitril/valsartan on the remodeling process.38) The study showed that sacubitril/valsartan was associated with reductions in NT-proBNP levels that persisted over a 12 month follow-up period in patients with HFrEF and with significant improvements in EF of 9.4 units and reductions in LV end-diastolic and end-systolic volume indices of 12.25 and 15.29 mL/m2, respectively. As shown in the Figure 1, patients with the largest reduction in NT-proBNP and left ventricular end-systolic volume index by 6 months had the lowest rates of death or HF hospitalization by 12 months, supporting the possibility that improved outcomes with sacubitril/valsartan were related to the reverse remodeling effects of the drug.
Figure 1
Relationship between NT-proBNP reductions, reverse remodeling and reduction in clinical events in the PROVE-HF study.
HF = heart failure; LVESVi = left ventricular end-systolic volume index; NT-proBNP = N-terminal pro-B-type natriuretic peptide.
The results of PARADIGM-HF and PIONEER-HF have strongly impacted guideline recommendations for the management of patients with HFrEF. Whereas RAAS inhibition remains a central pillar of therapy, use of an ARNI, rather than an ACE inhibitor or ARB, is now preferred. In the 2021 Update to the 2017 American College of Cardiology (ACC) Expert Consensus Decision Pathway for Optimization of Heart Failure Treatment further concluded that initiation of an ACE inhibitor or an ARB prior to switching is no longer required and patients can be started directly on an ARNI as the initial therapy.27) This document also provides specific recommendations about dosing as well as cautions and contraindications to the use of ARNIs. Similarly, the 2021 European Society of Cardiology (ESC) Guidelines for the treatment of HF give the use of an ARNI a class I recommendation stating that sacubitril/valsartan is recommended as a replacement for an ACE inhibitor in patients who remain symptomatic to reduce the risk of HF hospitalization and death and that de novo initiation without prior ACE inhibitor or ARB use may be considered.26)
Mineralocorticoid receptor antagonists
While Ang II is an important regulator of aldosterone, it is not the only pathway involved. There is evidence that circulating levels of this steroid mediator are not fully suppressed by either ACE inhibitors or ARBs.39) The association of aldosterone with sodium retention, potassium and magnesium loss, myocardial collagen production, ventricular hypertrophy, myocardial norepinephrine release and endothelial dysfunction made it an inviting therapeutic target and clinical trials provided evidence that mineralocorticoid receptor antagonists (MRAs) can improve outcomes in patients with HFrEF (Table 3). The Randomized Aldactone Evaluation Study (RALES) trial randomized HFrEF patients with severe NYHA class III/IV symptoms, 95% of whom were receiving an ACE inhibitor to receive either spironolactone 25 mg daily or placebo.40) The trial was stopped prematurely after an interim analysis found a 30% reduction in mortality risk (primary endpoint) with spironolactone. There was also a significant reduction in HF hospitalizations and HF symptoms in patients receiving active drug. Hyperkalemia was minimal in both treatment arms but gynecomastia was more prominent in patients receiving spironolactone, occurring in 10% compared to only 1% in patients receiving placebo. In Eplerenone Post–Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), patients with evidence of LV dysfunction and HF following an MI were randomized to receive either eplerenone 50 mg daily or placebo.41) All-cause mortality (primary endpoint) was significantly reduced by 15% and the co-primary end point of CV death or CV hospitalization was reduced by 13% with eplerenone. Serious hyperkalemia was more common with eplerenone compared to placebo (5.5% vs. 3.9%) while hypokalemia was less common.
Table 3
Mineralocorticoid receptor antagonist trials in HF with reduced EF
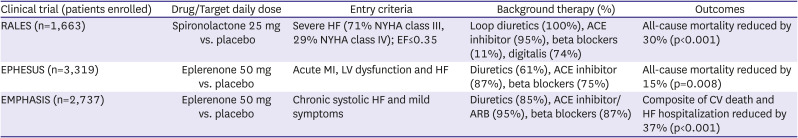
Whereas RALES and EPHESUS determined that MRAs improve outcomes in patients with either chronic severe HF or post-MI HF, whether they were effective in patients with milder HF was uncertain. The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS) trial42) randomized 2,737 patients with NYHA class II symptoms due to HFrEF, of whom 95% were receiving an ACE inhibitor or ARB, to eplerenone or placebo. The composite primary outcome of CV death or HF hospitalization was reduced by 37% in eplerenone treated patients due to significant reductions in death due to CV causes and in HF hospitalizations. Hyperkalemia was detected in 11.8% of eplerenone treated compared to 7.2% of placebo treated patients.
Based on the results of RALES, EPHESUS, and EMPHASIS (summarized in Table 3), MRAs receive a class I recommendation in HF management guidelines for treating symptomatic patients with HFrEF, provided that renal function and potassium levels are in acceptable range.25)26) Follow-up labs after initiation or uptitration of MRAs in required to assess effects on potassium levels and kidney function.
Beta blockers
Although the important role played by the SNS in the pathophysiology of HF had been recognized for many years,2)3)4) concerns about further compromising cardiac function by blocking the stimulatory effects of catecholamines delayed the testing of beta blockers in patients with HFrEF. It was not until the 1970’s when the concept that beta adrenergic blockade might be effective in patients with HFrEF began to take hold. An early publication assessing this possibility came from the pioneering group in Sweden of Waagstein et al.43) They reported that 7 patients with advanced HF and tachycardia who were treated with high dose beta blocker therapy over a period of 2–12 months experienced improved exercise capacity and cardiac function and a reduction in cardiac size. The first trial in HFrEF was the Metoprolol in Dilated Cardiomyopathy (MDC) trial, which randomized 383 patients with HFrEF and class II or III HF symptoms to metoprolol tartrate or placebo. Patients were required to demonstrate that they could tolerate low-dose metoprolol tartrate during a run-in period44) before dose was then gradually up-titrated to a target of 100–150 mg daily. There were 34% fewer primary endpoints (death or need for transplant) in the metoprolol group, an effect of borderline significance. Metoprolol therapy was associated with significantly greater improvement in EF and reduction in pulmonary artery wedge pressure than placebo. The MDC trial provided the first randomized clinical trial evidence of efficacy of beta blockers in patients with HFrEF and it solidified the practice of starting drug at a low dose with slow uptitration over time.
Encouraged by the MDC results, subsequent large-scale trials in patients with HFrEF (summarized in Table 4) were initiated during the 1990’s. The first to report was the US Carvedilol Clinical Trials Program which enrolled 1,094 patients with chronic HFrEF in a double-blind, placebo-controlled, stratified program.45) Patients were assigned to one of four treatment protocols based on their exercise capacity. They received either placebo or carvedilol in addition to background therapy, including an ACE inhibitor in 95%. The study was terminated prematurely when a mortality reduction of 65% in carvedilol treated patients was observed. Carvedilol was also associated with a 27% reduction in the risk of CV hospitalization and a 38% reduction in the combined risk of hospitalization or death. Worsening HF was less frequent with carvedilol than with placebo, an observation that helped dispel lingering fears that beta blockade would cause stable patients with mild to moderate HFrEF to deteriorate. The Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) study addressed the safety and efficacy of carvedilol in 2,289 patients with severe HF, defined as the presence of symptoms with minimal exertion and an EF <25%.46) Use of ACE inhibitor or ARB was nearly universal in the study population. COPENRICUS was also stopped prematurely when overwhelming evidence of benefit including a 35% reduction in all-cause mortality was observed with carvedilol. There was also a 24% risk reduction in combined risk of death or hospitalization. Overall withdrawal rates were significantly lower in the carvedilol than in the placebo group.
Table 4
Key clinical trials with beta blockers in HF with reduced EF
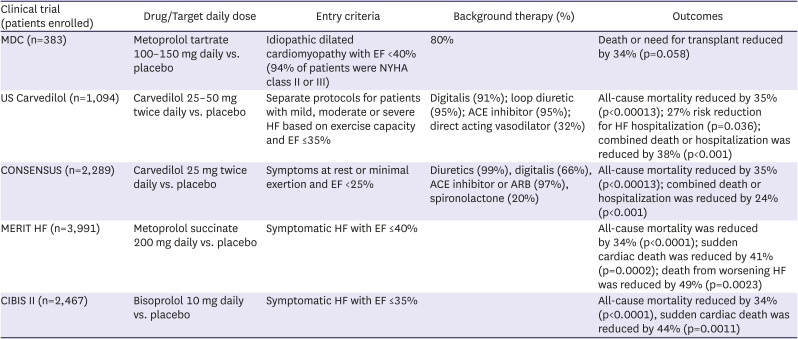
In Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF), 3,991 patients with chronic stable symptomatic HFrEF were randomized to either long acting metoprolol succinate, targeted at 200 mg daily or placebo.47) Again, the study was stopped prematurely on the recommendation of the DSMB which noted a 36% reduction with metoprolol in the primary endpoint of all-cause mortality. In CIBIS-II 2,647 symptomatic patients with NYHA class III or IV, symptoms and EF of 35% or less who were receiving standard therapy with diuretics and ACE inhibitors were assigned to either bisoprolol targeted at 10 mg daily or placebo.48)49) The study was stopped after a mean follow-up of 1.3 years when a significant 34% reduction in mortality risk was seen in the bisoprolol patients.
Based on results of these 4 trials, beta blockers now play a central role in guideline recommendations for managing patients with HFrEF. The beneficial effects of beta blockers in reducing mortality and hospitalization are additive to those of RAS inhibitors, as all of the trials required patients to be receiving an ACE inhibitor or ARB on entry. Unlike the situation with ACE inhibitors or ARBs, however, where improved outcomes were considered to be a class effect, recommendations for beta blockers specifically cite carvedilol, metoprolol succinate and bisoprolol, the three agents shown to be effective in the clinical trials. These agents differ from one another and from other beta blocking agents in their pharmacologic and pharmacokinetic properties. Carvedilol is a non-specific beta blocker with additional alpha blocking properties. It needs to be given twice daily to achieve therapeutic blood levels while metoprolol succinate and bisoprolol are both B-1 selective agents that can be administered once daily. These differences allow clinicians to individualize therapy according to patient characteristics. For instance, carvedilol is preferred when blood pressure is normal or elevated while metoprolol succinate or bisoprolol might be selected in patients with marginal blood pressure or those with reactive airway disease who might experience bronchoconstriction with B-2 blockade. Although patients with severe HFrEF were included in both COPERNICUS and CIBIS-II, beta blockers should be started in this population only at a time when patients are clinically stable. In all patients beta blockers are started at low dose and gradually uptitrated, with an increase in dose generally every 2 weeks until the target or maximally tolerated dose is achieved. Worsening fatigue and congestion can be seen during initiation or uptitration of beta blockers. Although these side effects usually resolve over the course of a few weeks as cardiac function improves, downtitration, increase in diuretics or much less commonly discontinuation of beta blockers may be required in some patients.
Sodium glucose cotransporter 2 inhibitors
Until recently, drugs used to treat diabetes mellitus (DM) had either a neutral effect on CV outcomes or were associated with increased risk.49)50) Sodium glucose cotransporter 2 (SGLT2) inhibitors are relatively new agents for treating patients with type 2 DM (T2DM). Their ability to treat hyperglycemia is based on the fact that virtually all glucose filtered by the kidney is reclaimed in the proximal tubule51) with the SGLT2 responsible for up to 90% of this reabsorption. Blocking the SGLT2 pathway reduces plasma glucose levels by increasing urinary loss. Additional SGLT2 inhibitor effects considered to be helpful in patients with T2DM include weight loss and reduction in blood pressure that is unaccompanied by increases in heart rate. Initial trial results assessing CV outcomes with the SGLT2 inhibitors empagliflozin, dapagliflozin and canagliflozin reported highly favorable effects, including impressive reductions in HF events.52)53)54) Moreover, the reduction in HF events occurred regardless of whether or patients had a diagnosis HF on entry into the trials.55)56) These findings raised the possibility that SGLT2 inhibitors might possess properties beyond their ability to lower glucose that could benefit patients with HF.51)57)
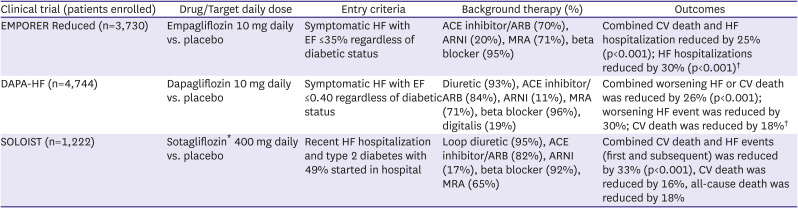
Based on results in patients with T2DM, a series of trials assessing the effects of SGLT2 inhibitors have now been completed in patients with HFrEF (Table 5). The Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure (DAPA-HF) trial randomized 4,744 patients with symptomatic HFrEF regardless of whether they had T2DM to dapagliflozin 10 mg once daily or placebo in addition to recommended HFrEF therapy.58) Over a follow-up of 18.2 months, there was a highly significant 26% reduction in the composite primary endpoint of worsening HF (defined as either hospitalization or urgent visit requiring intravenous HF therapy) or CV mortality. First worsening HF events were reduced by 30%, and CV and all-cause mortality were reduced by 18% and 17%, respectively. Benefits were similar in diabetic and non-diabetic patients. Neither volume depletion, renal dysfunction nor hypoglycemia differed between treatment groups. The Empagliflozin Outcome Trial in Patients with Chronic Heart Failure and a Reduced Ejection Fraction (EMPEROR-Reduced) included 3,730 symptomatic patients with HFrEF who were randomized to either 10 mg empagliflozin daily or placebo.59) During a median 16 month follow-up, the primary composite endpoint of CV death or HF hospitalization was reduced by 25% in the empagliflozin group, with consistent results in diabetic and non-diabetic patients. Patients receiving empagliflozin also experienced a 30% reduction in HF hospitalization and a significantly slower decline in estimated glomerular filtration rate (eGFR). Serious renal outcomes were also less frequent in the empagliflozin treated patients. Beneficial effects on renal function were seen in patients with and without chronic kidney disease and occurred regardless of baseline kidney function.60) Uncomplicated genital tract infections were more frequent with empagliflozin. A meta-analysis of DAPA-HF and EMPORER-Reduced demonstrated consistency between the results of the trials and supported the concept that dapagliflozin and empagliflozin reduce all-cause and CV death and improve renal outcomes in patients with HFrEF.61)
Table 5
SGLT2 inhibitor trials in HF with reduced EF
ACE = angiotensin converting enzyme; ARB = angiotensin receptor blocker; ARNI = angiotensin receptor neprilysin inhibitor; CV = cardiovascular; EF = ejection fraction; HF = heart failure; MRA = mineralocorticoid receptor antagonist; NYHA = New York Heart Association; SGLT = sodium glucose cotransporter.
*Sotagliflozin is a combined SGLT1 and SGLT2 inhibitor; †Benefits seen regardless of diabetic status.
While the results of DAPA-HF and EMPORER-Reduced demonstrate the safety and efficacy of SGLT2 inhibitors in stable outpatients with HFrEF, the effects of drug initiation during an episode of decompensated HF were unknown. In the Effect of Sotagliflozin on Cardiovascular Events in Patients with Type 2 Diabetes Post Worsening Heart Failure (SOLOIST-WHF) trial, 1,222 patients with T2DM who were recently hospitalized for worsening HF were assigned to either sotagliflozin (which inhibits SGLT1 in the bowel as well as SGLT2 in the kidney) or placebo.62) The primary endpoint was total number of CV deaths and HF events (hospitalizations or urgent visits). Although the trial was stopped prematurely due to funding issues, patients randomized to sotagliflozin experienced a highly significant 33% reduction in primary endpoint events and reductions of 16% and 18% in CV and all-cause mortality. Benefits were seen whether therapy was started in-hospital or shortly after discharge. Neither hypotension nor acute kidney injury were increased but diarrhea was more common in the sotagliflozin group, occurring in 6.1% of patients. The results of the EMPULSE study which assessed initiation of empagliflozin in patients during HF hospitalization were presented in November, 2021.63)64) In this study, 530 patients were randomized to either empagliflozin 10 mg or placebo during a HF hospitalization and then followed for up to 90 days after discharge. Although EF was not part of the entry criteria, median value in the study population was 31%. The stratified win ratio, defined as a composite of death, number of HF events, time to first HF event, and change in Kansas City Cardiomyopathy Questionnaire-Total Symptom Score (KCCQ-TSS) from baseline to 90 days demonstrated a significantly greater likelihood of clinical benefit in the empagliflozin group (53.9%) compared with the placebo group (39.7%).
The SGLT2 inhibitors provide an exciting new approach for treating patients with HFrEF. Although effective as hypoglycemic agents, it is clear that SGLT2 effects on CV disease and HF, in particular, transcend their hypoglycemic properties and a variety of mechanisms have been postulated for their CV effects (Table 6). The independence of SGLT2 inhibitors from neurohormonal drugs, both in mechanism of action and side effect profile, enhances their usefulness as a HF therapy. Specifically, the absence of effects on heart rate, serum potassium levels, angioedema and only mild blood pressure lowering coupled with favorable effects on renal function helps facilitate the integration of the SGLT2 inhibitors in a drug regimen that includes an ARNI, MRA and beta blocker. Similarly, their effectiveness across a broad spectrum of the HF population regardless of diabetic status, age, LVEF, NYHA class, renal function or co-morbidities enhances their usefulness. Of particular note is that the benefits of SGLT2 inhibitors appear to be additive to those of other guideline directed medical therapies (GDMTs) supporting the concept that they should be used in combination rather than as replacement for another agent. Side effects associated with SGLT2 inhibitors include hypotension, urinary tract infection, genital mycotic infections (i.e., Fournier’s gangrene), diarrhea, bone fractures and adverse events leading to limb amputation.
Table 6
Possible mechanisms by which SGLT2 inhibitors decrease the severity of heart failure
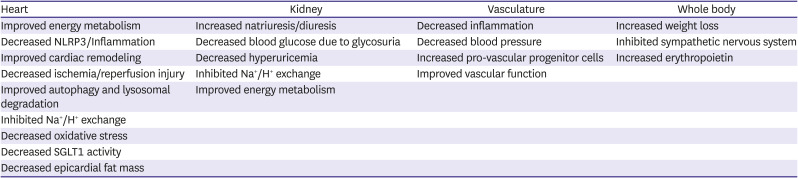
From: Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: a state-of-the-art review. JACC Basic Transl Sci 2020;5:632-44.57)
NLRP3 = nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain-containing 3; SGLT2 = sodium glucose cotransporter 2.
Based on clinical trial results described above, SGLT2 inhibitors have now assumed an important role in treating patients with HFrEF. The 2021 ESC Guidelines provided a class I, recommendation, based on LOE A for the use of empagliflozin and dapagliflozin to reduce the risk of HF hospitalization and death in all patients with symptomatic HF (i.e., NYHA class II–IV) and LVEF ≤0.40.26) The 2021 Update to the 2017 ACC Expert Consensus Decision Pathway for Optimization of Heart Failure Treatment recommend that either empagliflozin or dapagliflozin be added to an ACE inhibitor, ARB or ARNI (preferred), an evidence based beta blocker and diuretics (as needed) in patients with symptomatic HF.27) The document further recommends use in patients with eGFR ≥20 mL/min/1.73 m2 for empagliflozin and ≥30 mL/min/1.73 m2 for dapagliflozin, based on clinical trial entry criteria. Guidance for the use of SGLT2 inhibitors is certain to evolve as further trial results become available and clinical experience grows. In particular, based on the results of SOLOIST-WHF and EMPULSE, recommendations about in-hospital initiation of SGLT2 inhibitors are anticipated.
AGENTS CONSIDERED IN ADDITION TO OR AS ALTERNATIVES TO CORE THERAPY
Hydralazine and isosorbide dinitrate combination
The use of hydralazine in combination with a long acting nitrate was developed in the 1970’s based on early studies showing that substantial hemodynamic improvement could be obtained by unloading the failing heart.65)66)67) This treatment was tested in the Vasodilator Heart Failure Trial (V-HeFT) in which 642 male patients were assigned to either the hydralazine/isosorbide dinitrate combination at doses of 300 and 160 mg daily, respectively, prazosin at 20 mg daily or placebo in addition to background therapy, which at the time of the trial consisted predominantly of digoxin and diuretics.68) A borderline significant reduction in mortality with the hydralazine/isosorbide nitrate combination compared to placebo was observed over the average 2.3 year follow-up period. For the pre-specified endpoint of mortality by two and three years, risk reduction with hydralazine/isosorbide dinitrate was 34% and 36%, respectively. An increase in EF was seen only in the hydralazine/isosorbide dinitrate group. In V-HeFT II, a study that included 804 men, hydralazine/isosorbide dinitrate at the same dose as in V-HeFT I was compared to enalapril at 20 mg daily.69) Despite a greater improvements in EF and peak VO2 during exercise with the hydralazine/isosorbide combination, there was a 28% reduction in the risk for all-cause mortality with the ACE inhibitor.
Based on the robust data base demonstrating efficacy of ACE inhibitors from studies summarized in Table 2, the superiority of enalapril in V-HeFT II, practical issues related to the thrice daily dosing requirement of hydralazine/isosorbide dinitrate and relatively high incidence of troublesome side effects such as headache, gastrointestinal disturbances and hypotension, the hydralazine/isosorbide combination is now recommended for patients who are unable to tolerate either an ARNI, ACE inhibitor or ARB due to advanced kidney disease or side effects. An exception is in Black patients with HFrEF in whom there is evidence that the hydralazine/isosorbide dinitrate combination significantly improved outcomes, including a 43% reduction in the risk of death.70) As a result, guidelines recommend consideration of the hydralazine/isosorbide dinitrate combination in Black patients receiving core therapies who remain symptomatic.
Ivabradine
Resting heart rate is an indicator of mortality risk in patients with CV disease.71)72) Hyperpolarization-activated, cyclic nucleotide-gated cation currents, termed I(f) channels play an important role in pacemaker function in the heart.73) Ivabradine, a drug that specifically inhibits the I(f) current in the sinoatrial node, can lower heart rate without affecting other aspects of cardiac function.74)75) In the ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL) trial, patients with stable coronary artery disease and systolic dysfunction, most of whom were already receiving beta blockers, were assigned to either placebo or 7.5 mg daily of ivrabadine.76) Ivabradine reduced heart rate by an average of 6 beats/minute, but failed to significantly reduce the primary study endpoint of CV death, MI, and HF hospitalization in either the entire population nor in patients with heart rate above 70 beats/minute. It did, however, reduce secondary endpoints of admission to hospital for fatal and non-fatal MI by 36% and coronary revascularization by 30%. In Study Assessing the Morbidity–Mortality Benefits of the I
f Inhibitor Ivabradine in Patients with Coronary Artery Disease (SIGNIFY), conducted in 19,102 patients with stable coronary artery disease without clinical HF and a heart rate of ≥70 beats/ivabradine at a dose of 10 mg daily failed to improve a composite outcome of CV death or nonfatal MI.77)
The effect of heart-rate reduction with ivabradine at a dose of 7.5 mg on outcomes in HF patients was assessed in the Systolic Heart Failure Treatment with the I(f) Inhibitor Ivabradine Trial (SHIFT) trial, a study of 6,558 patients with symptomatic HF and a LVEF of 35% or lower, sinus rhythm with heart rate 70 beats/minute or higher, a HF hospitalization within the previous year who were on stable background treatment including a beta blocker, if tolerated.78) The primary composite endpoint of CV death or HF hospitalization was reduced by 18% due primarily to 26% reductions in both HF hospitalizations and deaths due to HF. Although fewer serious adverse events occurred with ivabradine than with placebo, patients receiving ivabradine experienced more symptomatic bradycardia and visual side-effects (phosphenes) than did those randomized to placebo. An increased incidence of atrial fibrillation has also been reported with ivabradine.79)
Based on the findings of SHIFT, ESC guidelines recommend that ivabradine should be considered in symptomatic patients with HFrEF in sinus rhythm with a heart rate 70 or above beats/minute who are on optimal tolerated doses of guideline directed medical therapies (or demonstrated intolerance) including an evidence-based beta blocker to reduce the risk of HF hospitalization or CV death.
Vericiguat
Soluble guanylate cyclase (sGC) is a second messenger that internalizes messages carried by peptide hormones and is the most sensitive physiologic receptor for nitric oxide (NO) within cells.80)81)82)83) Binding of NO to the heme moiety of sGC induces synthesis of the second messenger cyclic guanosine monophosphate (cGMP). The sGC pathway is an important regulator of CV function including vasodilation. In patients with HF, endothelial cell dysfunction and increased levels of reactive oxygen specific reduce NO bioavailability, thereby impairing cGMP production. Vericiguat is an oral soluble guanylate cyclase stimulator that directly stimulates sGC through a binding site independent of NO.84) It also sensitizes sGC to endogenous NO by stabilizing NO binding to the binding site on the enzyme. Unlike nitrate preparations, vericiguat does not cause oxidative stress or induce endothelin-1 production, nor does it develop tolerance. Initial testing of vericiguat in a phase 2b dose-finding trial involving patients with worsening high-risk HF and a reduced EF, reported that higher doses or vericiguat were associated with a significant reduction in NT-proBNP, a biomarker which has been shown to be predictive of clinical outcome.85) Rates of adverse events were similar between the two study groups suggesting that it could be administered safely when gradually up-titrated.
In the Vericiguat Global Study in Subjects with Heart Failure with Reduced Ejection Fraction (VICTORIA), 5,050 high risk patients with chronic symptomatic HFrEF, EF <45% and recently decompensated HF were randomized to either vericiguat titrated to 10 mg daily or placebo, in addition to standard medical therapy.86) The primary outcome was a composite of death from CV causes or first HF hospitalization. Due to the high-risk profile of the study population, clinical events occurred early so that the pre-defined number of endpoint events was reached after a mean patient follow-up period of only 10.8 months. Nonetheless, the primary endpoint was significantly reduced by 10% in the vericiguat group, an effect driven primary by a reduction in HF hospitalization. Anemia was more common in patients who received vericiguat. Hypotension was equally common in the study groups and there were no significant differences between them in the rates of symptomatic hypotension or syncope. Subgroup analysis of the VICTORIA results demonstrated that vericiguat did not reduce the primary endpoint in patients in the highest quartile of entry NT-proBNP levels (>5,314 pg/mL) but was associated with a 23% risk reduction in patients with levels ≤4,000 pg/mL,87) suggesting that the clinical course of patients with severe HF (manifest by extremely high levels of natriuretic peptides) will not be improved by vericiguat. Although the 2021 ESC Guidelines recommends that vericiguat may be considered in addition to core therapy for HFrEF to reduce risk of CV mortality and HF hospitalization, additional clinical trials that are being planned should help solidify future recommendations, particularly in regards to the target population.
Digoxin
For over 2 centuries, digitalis glycosides were a mainstay of therapy for HF.88)89)90) With the availability of more effective drugs they are now used considerably less frequently than in the past. Digoxin, originally derived from digitalis leaf, is the most common used glycoside in the family. Its acts primarily to inhibit the sodium potassium adenosine triphosphatase (Na+/K+ ATPase), mainly in the myocardium.91) The resultant increase in intracellular calcium increases myocardial contractility, stroke volume and blood pressure while reducing heart rate. Digoxin also has parasympathetic effects that can slow conduction through the atrioventricular node making it a useful adjunct for slowing heart rate in patients with atrial fibrillation. In the Digitalis Investigation Group (DIG) trial there was a trend towards a reduction in mortality and a significant reduction in both overall and HF hospitalization in patients receiving digoxin.91) The 2021 ESC guidelines recommend that digoxin may be considered in patients with HFrEF in sinus rhythm and may be useful for the treatment of patients with HFrEF and atrial fibrillation with rapid ventricular response when other therapies cannot be pursued. Digoxin is cleared by a renal mechanism and blood levels rise as renal dysfunction worsens. Caution is recommended when using in females, older or frail patients and those who are hypokalemic or malnourished. When digoxin is used, levels should be checked and maintained <1.2 ng/mL as higher levels have been associated with toxicity, including arrhythmias.92)93)
NEWER AGENTS
Omecamtive mecarbil
Omecamtiv mecarbil is the first of a new class of drugs, termed myotropes94) which stabilize the pre-power-stroke state of myosin, thereby enabling more myosin heads to undergo a power-stroke during systole.95)96)97) The increased ‘number of hands pulling on the rope’ produces more force during each cycle of cardiac contraction. A potential downside is that coronary filling, which occurs predominantly during diastole, could be compromised by sustaining the interaction between the myofilaments and lengthening systolic ejection time. Preliminary studies with omecamtiv mecarbil identified blood levels that could improve myocardial contractility without reducing coronary blood flow.98) Using pharmacokinetic guidance to adjust drug dose, administration of omecamtiv mecarbil to levels shown to increase myocardial contractility were well tolerated during exercise in patients with ischemic cardiomyopathy and a history of angina, suggesting that this strategy could protect against myocardial ischemia in clinical practice.99) In the pilot Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF) patients were randomly assigned to receive 25 mg oral omecamtiv mecarbil twice daily (fixed-dose group), 25 mg twice daily titrated to 50 mg twice daily guided by pharmacokinetics (pharmacokinetic-titration group), or placebo for 20 weeks.100) Higher plasma levels were reached with pharmacokinetic-titration than with fixed-dose administration and there were significantly greater increases in systolic ejection time and stroke volume and decreases in LV end-diastolic diameter, heart rate and NT-proBNP levels in the pharmacokinetic-titration group than in the placebo group.
In the phase III Global Approach to Lowering Adverse Cardiac outcomes Through Improving Contractility in Heart Failure (GALACTIC-HF) trial, 8,256 patients with symptomatic chronic HFrEF were enrolled from both in- and out-patient settings and followed over a median of 21.8 months after randomization to pharmacokinetic guided doses of 25, 37.5 and 50 mg of omecamtiv mecarbil or placebo.101) Risk for the composite primary outcome of first HF event or CV death was significantly reduced by 8% in patients randomized to omecamtiv mecarbil compared to placebo, a result due predominantly to a reduction in HF events.102) Omecamtiv treatment was also associated with a 10% reduction in levels of NT-proBNP. Small increases in cardiac troponin I level were not accompanied by an increase in ventricular arrhythmia or ischemic events. Post-hoc analysis of the results of GALACTIC-HF in the subgroup of patients with severe HF defined as NYHA class III to IV, LVEF ≤30%, and hospitalization for HF within the previous 6 months, showed a significantly better effect of the drug on the primary study outcome than in patients not meeting severe HF criteria, suggesting that omecamtiv mecarbil may have greater utility in more advanced HFrEF patients.103) Consideration of the use of omecamtiv mecarbil in future guideline recommendations is anticipated, perhaps as adjunctive therapy in patients with more severe disease and lower baseline EF.
Gene and cell therapies
After a period of relative dormancy, exploration of gene and cell therapies for HFrEF has picked up momentum in recent years. As pivotal clinical trials have not yet been carried out for either, this section will provide a brief overview of these therapies and the early results from selected studies in each.
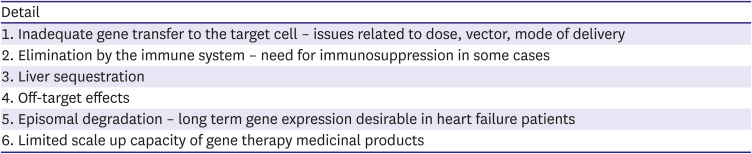
Gene therapy for cardiac disease involves delivery of genetic material (either DNA or RNA) or editing existing genes (using CRISPR/Cas9-based genome engineering) in order to modify production of molecules that influence cardiac structure and function. Although molecules that are deficient in the failing heart are the most common targets, reducing gene expression may be desirable in certain conditions. Expression of a variety of enzymes, receptors, signaling molecules and structural proteins that are dysregulated in either acquired or monogenetic disease are potential targets for gene therapy.104)105) Important issues that need to be considered for successful gene therapy (summarized in Table 7) include delivering adequate amounts of genetic material to the heart, minimizing side effects and uptake in other organs and treating the immune response to either the vector or the gene product, itself.
Table 7
Potential pitfalls of gene therapy for heart failure
Although a variety of vectors and approaches can be used to in gene therapy of HF, recent studies have focused on gene delivery using viral vectors that are introduced into the systemic or coronary circulation. Either adenoviruses or adeno-associated viruses (AAV) are preferred. One early study used an AAV type 1 serotype vector to deliver copies of the human SERCA2a gene to the myocardium of patients with acquired HFrEF. The SERCA2a enzyme plays a crucial role in regulating intracellular calcium flux in cardiomyocytes and there is evidence that it is deficient in in patients with HFrEF.106)107) Studies in isolated cells and experimental animal models showing that viral based SERCA2a gene therapy could rescue the HF phenotype were the basis of the Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) studies in which AAV1/hSERCA2a was administered to patients with HFrEF. Despite success in phase 1 and 2 studies using this approach,108)109) the pivotal CUPID-2 study failed to demonstrate evidence of clinical benefit.110) Whether this was related to selection of SERCA2a as the target, inability to deliver adequate copies of the SERCA2a gene to the myocardium or other issues remains uncertain as the program was terminated shortly after the results of CUPID-2 became available.
Monogenetic disorders can cause both dilated and hypertrophic cardiomyopathy. Danon disease is caused by a mutation in the LAMP2b gene that results in impaired autophagy and buildup of debris in cells throughout the body.111)112) Cardiac effects are prominent with hypertrophic cardiomyopathy seen in males and either dilated or hypertrophic cardiomyopathy appearing in females. Survival is greatly reduced, particularly in males, most of whom die in the second or third decade of life. An AAV9/LAMP2b construct was shown to reverse many of the abnormalities in myocardial structure in a rodent model of Danon disease. Intravenous delivery of AAV9/LAMP2b to 5 adolescent or young adult male patients increased LAMP2b protein expression in the myocardium for at least 12 months, improved myocardial architecture, reduced the quantity of intracellular vacuoles that are characteristic of the disease and was associated with clinical stabilization.113) Adverse effects of the treatment and the associated immunosuppression that was required, albeit severe in some cases, were manageable and all have resolved over time. If these early findings are confirmed in additional patients, gene therapy will provide a potential treatment for a disease for which no other cure exists.
There is also ongoing work in the development of cell therapy to treat patients with HF. Most of this work involves stem cells or induced pluripotent stem cells (iPSCs) which are generated directly from a somatic cell. A variety of cells have been used including those derived from the heart, bone marrow and other sources. Mechanisms for improved cardiac function include replacement or generation of cardiomyocytes, growth of new blood vessels and secretion of autocrine or paracrine growth factors which can effect cells in the heart. The field has been controversial and to date, convincing evidence of improved outcomes with any form of cell therapy for patients with HF is lacking. Results from the DREAM-HF study which was presented at the 2021 American Heart Association (AHA) meeting (but not yet published) are promising.114) In this trial 537 patients with HFrEF due to ischemic or non-ischemic etiology were randomly assigned to received 150 million allogeneic mesenchymal precursor cells (MPCs) injected directly into areas of the myocardium deemed to be viable based on electrode mapping. Although the study failed to improve HF outcomes, there were significant reductions in irreversible CV events defined as MI or stroke which appeared to be more substantial in patients with higher levels of the inflammatory marker, C-reactive protein (CRP). While the findings need to be confirmed in future studies, the results suggest that MPCs benefit patients with HFrEF by reducing inflammation and generating new vessels in the heart.
IMPLEMENTATION OF EVIDENCE BASED THERAPIES IN THE HEART FAILURE WITH REDUCED EJECTION FRACTION POPULATION
One of the most vexing problems in the management of HFrEF is the underutilization and underdosing of GDMT in a high percentage of patients. The Change the Management of Patients with Heart Failure (CHAMP-HF) registry which included 3,518 patients from primary care and cardiology practices in the US, reported that GDMT using ACE inhibitor/ARB/ARNI, beta blocker, and MRA was not being used in 27%, 33%, and 67% of eligible patients, respectively.115) Moreover, target doses of each these three classes of drugs were reached in only 14–28% of the patients. Astonishingly only 1% of patients who were eligible for all classes of medication were simultaneously receiving target doses of drugs of each of the 3 classes. Follow-up of a subset of the population revealed that few patients had doses increased over time.116) Underutilization and underdosing were associated with older age, lower blood pressure, more severe functional class, renal insufficiency, and recent HF hospitalization.
While data relating dose to clinical benefit for GDMTs of HFrEF is not particularly robust, trials with ACE inhibitors and beta blockers have demonstrated significantly better outcomes when target dose is reached.117)118)119) In some cases, underdosing is related to drug related side effects, but it is often due to other factors including provider or patient reluctance, theoretical concern about potential side effects, logistical considerations related to the need for additional contact between patient and provider and requirement for additional lab evaluation that may be needed when some drugs (e.g., RAS inhibitors and MRAs) are uptitrated. Development of limiting side effects is a valid reason for not uptitrating dose of GDMT and should be differentiated from these other considerations. When dose limiting side effects are present, maintaining patients on the dose of drug that is tolerated is advised as therapeutic efficacy appears to be maintained.120) The use of GDMT is often lowest in older patients despite evidence that efficacy of core drugs is not compromised by age.121) In the Korean Acute Heart Failure (KorAHF) registry of 2,045 patients age 65 years or older, use of GDMT decreased with advancing age despite the fact that it was associated with a significant reduction in all-cause mortality in the old and very elderly patients aged ≥80 years.122)
Given the enormous opportunity to improve outcome in patients with HFrEF by using the core and supplemental therapies outlined in this review, greater attention to implementation of HF therapies and development of effective public health strategies to achieve that goal is clearly warranted. Even as new approaches are being developed, we can greatly improve the quality of life and reduce morbidity and mortality in patients with HFrEF by utilizing drugs that are now available.123)
 XML Download
XML Download