

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Myelofibrosis (MF) is a myeloproliferative neoplasm characterized by bone marrow fibrosis, progressive splenomegaly, cytopenia, constitutional symptoms including weight loss, fever, and night sweats. It is associated with an increased risk of early death due to secondary leukemic transformation, infections, bleeding, progressive cachexia, and cardiovascular events [1].
The International Prognostic Scoring System (IPSS) at diagnosis and dynamic IPSS (DIPSS) during the disease were developed by the International Working Group for Myelopro-liferative Neoplasm Research and Treatment (IWG-MRT) [2]. DIPSS-plus was modified with the addition of platelets count <100×109/L, transfusion need and unfavorable karyotype features to DIPSS. The DIPSS-plus also stratifies patients into four risk groups: low-risk, intermediate -1 risk (INT-1), intermediate-2 risk (INT-2), and high-risk group with respective median survival rates of 15.4, 6.5, 2.9, 1.3 years [3]. Other prognostic models include mutation-enhanced IPSS for patients with PMF age ≤70 years (MIPSS70) [4], MIPSS70+ version 2.0 [5], genetically inspired prognostic scoring system (GIPSS) [6], incorporating cytogenetic information and mutational status.
There are various treatment options for palliative and supportive treatment. Although allogenic stem cell transplantation is the only curative treatment, it carries a high risk of morbidity and mortality [7]. Ruxolitinib, a Janus kinase-1 and 2 (JAK1/2) inhibitor, is a treatment option to relieve symptomatic splenomegaly and constitutional symptoms in patients with INT-2 and high-risk MF. Spleen response rates were reported as 41.9% at 24 weeks and 53.4% at any time in the COMFORT I and II studies, respectively. In addition, constitutional symptoms improved significantly in both the studies [8, 9]. The most common side effects were thrombocytopenia and anemia. However, these studies did not include patients with MF or INT-1 risk. According to recent literature, the JUMP study showed that ruxolitinib had significant clinical benefits with acceptable tolerability in INT-1 risk MF patients with splenomegaly [10].
In this retrospective study, we aimed to document real-life clinical experiences with ruxolitinib in primary and secondary MF patients.
MATERIALS AND METHODS
Patients
This was a single-institution retrospective cohort of patients with primary MF (PMF), post-polycythemia vera MF (post-PV MF), and post-essential thrombocythemia MF (post-ET MF), which were diagnosed according to World Health Organization (WHO) 2016 criteria [11].
Fifty-seven patients who received ruxolitinib were included in the analyses. All patients were at least 18 years of age and had INT-1/2 or high-risk disease according to the DIPSS-plus risk classification. Ruxolitinib was used for symptomatic disease and/or symptomatic splenomegaly, which is resistant or intolerant to conventional cytoreductive therapy in patients with IPSS INT-1 and INT-2, or high risk. Ruxolitinib doses that were administered ranged from 5–25 mg twice daily. Dose modifications were made according to efficacy and side effect profile as previously described [8]. All data including demographic characteristics, laboratory test results, ultrasound reports, symptom assessment questionnaires, survival status, and side effect profile were recorded from patient’s chart file. Approval was obtained from the institutional ethics board.
Definitions
Spleen size was measured by ultrasound examination. Myeloproliferative neoplasm symptom assessment from the total symptom score (MPN-SAF TSS), a disease-specific questionnaire, was used to evaluate the symptom response. Symptom response was defined as at least a 50% reduction in myelofibrosis-related symptoms. A symptom improvement of 5% to 49% was termed clinical improvement.
Overall survival (OS) was estimated as the time elapsed from the beginning of ruxolitinib until death of any cause or last contact. Common Terminology Criteria for Adverse Events (CTCAE) version 5 were used to grade the side effects.
Endpoints
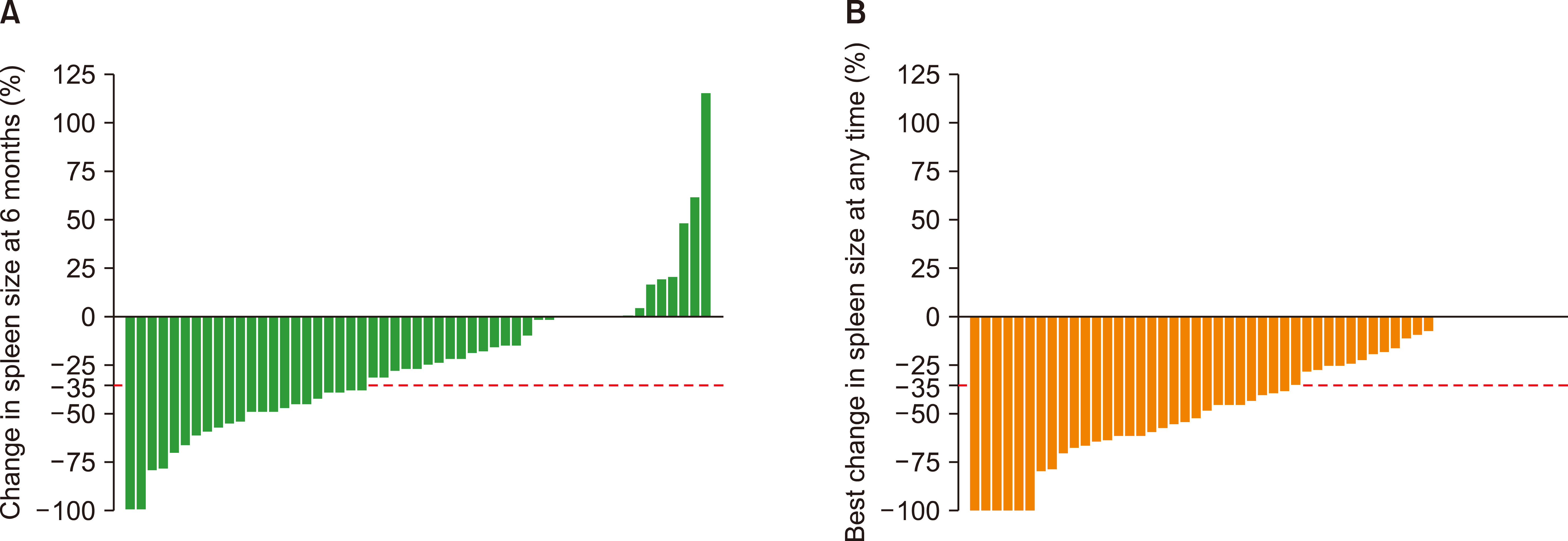
The primary endpoint was spleen response, which was defined as at least a 35% reduction in spleen size at any time during the therapy. Secondary endpoints included spleen response at the sixth month of therapy, symptom response, clinical improvement, overall survival, and safety profile of ruxolitinib.
Statistical analyses
Data normality was assessed using the Shapiro–Wilk test. Normally distributed continuous data were expressed as the mean±standard deviation. Skewed data were reported as the median (range). Survival probability was estimated using the Kaplan–Meier method. Survival comparisons were performed using the log-rank test. Statistical significance was set at P≤0.05. All analyses were performed using Stata/MP 14.1 for Mac (64-bit Intel) (TX, USA).
RESULTS
Patients
Fifty-seven patients with MF who received ruxolitinib were included in this study. The median age of the patients in this study cohort was 58 (23–80) years. Overall, 50.9% of the patients were male, and 49.2% had PMF. Patient assignment according to DIPSS-plus risk categories was as follows: 33 patients (57.9%) in INT-1, 23 patients (40.4%) in INT-2, and 1 patient (1.8%) at high risk. IPSS categorized 56.1% of patients in INT-1 and 43.9% in INT2/high-risk patients. Hydroxyurea was used previously in 91.2% of patients before ruxolitinib. JAK2 was positive in 71.9% of patients. Splenectomy was performed in only four (7%) patients (Table 1).
Efficacy
Since four patients underwent splenectomy, a total of 53 patients were available for spleen response. Overall, spleen size reduction of at least 35% (spleen response) was achieved in 56.6% of patients (30/53) at any time. A spleen response at 6 months was observed in 43.4% of patients (Fig. 1). In contrast, 19 patients with INT-1 risk disease (63.3%) responded at any time during follow-up. Only 43.3% (13/30) of the patients in the INT-1 risk group achieved a spleen response at 6 months. The best and 6-months spleen response rates for INT-2/high-risk patients were 47.8% (11/23) and 43.5% (10/23), respectively.
Twenty-three patients had the MPN-SAF TSS during the course of treatment. Five patients (21.7%) had at least a 50% reduction (symptom response) in myelofibrosis-associated symptoms. In 10 patients (43.4%), a 20-50% reduction in symptom scores was observed. Four patients (17.3%) had only mild (5–10%). Four patients did not respond.
Safety
The median duration of exposure to ruxolitinib was 22 months (range, 1–80 mo). Forty-two patients (73.7%) still received ruxolitinib. The initial dose of ruxolitinib was 20 mg bid in 67% of patients. The dose of ruxolitinib was adjusted according to side effects and efficacy. Most of the patients continued to use the usual doses of ruxolitinib during follow-up (Supplementary Fig. 1). The discontinuation rate was approximately 26.3%.
Fifty-two out of 57 (91%) patients experienced hematologic side effects. The most common hematologic adverse events were anemia (all grades, 84.2%; grade 3–4, 42.1%) and thrombocytopenia (all grades, 38.6%; grade 3–4, 17.5%; Table 2). Cytopenia was more prominent within the first three months. However, those tended to recover at the end of six months and approached near-normal values by the end of the year (Supplementary Fig. 2). These adverse events were mostly manageable, and only four patients discontinued treatment due to cytopenia.
A total of 27 out of 57 patients experienced non-hematologic side effects, which were generally grade 1–2. The most common non-hematologic adverse events were fatigue, pneumonia, and elevated liver enzyme levels. Pulmonary embolism was observed in two patients who had pneumonia. Herpes zoster infection, hepatitis B virus (HBV) reactivation, and urinary tract infection were noted. Unexpected adverse events included peripheral polyneuropathy, cataracts, proteinuria, and pancreatic adenocarcinoma. Treatment was discontinued because of these adverse events. Overall, three patients were diagnosed with cancer, including pancreatic adenocarcinoma, squamous cell carcinoma of the skin, and metastatic neuroendocrine tumor (Table 2). Patients with INT-1 risk had a similar safety profile (Supplementary Table 1).
Survival
A total of 17 (29.8%) deaths occurred during a median follow-up period of 22 months. The causes of death were as follows: transformation to acute myeloid leukemia (N=5), disease progression (N=11), and pancreatic adenocarcinoma (N=1). In all cohorts, the OS probability at 24 months was 75.1% (95% CI, 59.8–85.3%). The median overall survival was not reached (Fig. 2A).
Median survival was not reached for patients with INT-1 and INT-2 risk. Two-year OS probability was approximately 84.5% (95% CI, 63.1–94.0%) and 62.3% (95% CI, 37.5–79.6%), respectively (Fig. 2B).
Since previous studies claim that basal hemoglobin levels lower than 10 g/dL were associated with worse survival outcomes, we stratified survival probabilities according to basal hemoglobin levels [3, 12, 13] Median survival was not reached in both groups with Hb <10 g/dL and Hb ≥10 g/dL during the first 2 years, and patients with Hb ≥10 g/dL seem to have a better survival. Two-year survival probabilities were 58.5% (95% CI, 34.5–76.4%) versus 87.5% (95% CI, 65.5–95.9%), respectively. However, survival curves merged at 33 months and beyond (Fig. 2C, P=0.15).
DISCUSSION
In this study, we observed that ruxolitinib use led to a spleen and symptom response in approximately 56.6% and 21.7% of patients with MF, respectively, according to the 2013 IWG-MRT and European Leukemia Net (ELN) response criteria [14]. Hematological side effects were observed in almost all patients. However, these were mostly manageable. The non-hematologic side effects were generally mild.
The patient population in our cohort was younger than those reported in the COMFORT, JUMP, and UK ROBUST trials [8-10, 15]. Median ages in the abovementioned studies were 58, 66, 68, and 70 years, respectively. This is due to the fact that our cohort included more patients with INT-1 risk MF (33/57 patients, 57.9%) in comparison to JUMP (163/1144 patients, 14.2%) and UK ROBUST (14/48 patients, 29.2%) [10, 15]. The median ages in the INT-1 and INT-2/high-risk groups were 58 and 64 years, respectively, in our cohort. Consistently, the median age in the INT-1 risk group was 62 years in the JUMP study. Therefore, it should be considered that our results were mostly derived from a younger cohort with a lower-risk disease.
A spleen size reduction of at least 35% was achieved in 43.4% at 6 months in our population, which was similar to that of COMFORT-I (41.9%). Consistent with the COMFORT-II trial (53.4%), spleen response, which was achieved at any time during the follow-up, was approximately 56.6% [8, 9]. We found that the spleen response was approximately 63.3% and 43.3% in the INT-1 risk group at any time and sixth month of therapy, respectively. At 48 weeks, a ≥50% reduction in palpable spleen length was observed in 57.1% of patients with INT-1 risk in UK ROBUST [15] and 60.5% in the JUMP study [10]. In an independent study that evaluated the efficacy and safety of ruxolitinib in 70 patients in the INT-1 risk group, the spleen response was 54.7% at 6 months [16]. Our results confirm the efficacy of ruxolitinib in both INT-1 and higher risk groups. The presence of ASXL1, EZH2, SRSF2, U2AF1, or IDH1/2 mutations was defined as high molecular mutation (HMR). Unfortunately, the molecular risk profile cannot be documented in unresponsive patients. Two unresponsive patients in our cohort had non-cirrhotic portal hypertension.
Twenty-three patients underwent a symptom assessment. Most patients who received ruxolitinib showed an improvement in symptom scores. Overall, symptom responses and clinical improvements were observed in 21.7% and 60.7% of patients, respectively. In INT-1 patients, symptom response and clinical improvement were achieved in 60% and 40% of 10 evaluable patients, respectively. Although the symptom assessment scales vary across the studies, the symptom response rates were 45.9% in the COMFORT-I and 20.8% in the UK ROBUST studies, respectively [9, 15]. In the INT-1 risk group analyzed in the UK ROBUST study, the symptom response rate was approximately 21.4% [15]. However, a study reported a symptom response of up to 80% in 65 patients as well [16]. Therefore, our findings are consistent with previous reports regarding symptom evaluation.
Anemia and thrombocytopenia are common adverse effects. Grade 3–4 side effects occurred in 42.1% and 17.5% of patients, respectively. However, these adverse effects rarely lead to treatment discontinuation. It is well known that ruxolitinib treatment is frequently associated with anemia and thrombocytopenia in patients with myelofibrosis. In previous studies, grade 3–4 anemia and thrombocytopenia ranged from 22.5% to 45.2% and thrombocytopenia ranged from 0–15.2%, respectively. Anemia and thrombocytopenia tended to improve by the third month and reached a new steady state by the sixth month of therapy [8, 9, 17]. Our real-life observations are consistent with the findings of previous studies.
Palandri et al. [16] found that ruxolitinib-related anemia and thrombocytopenia were evident in 45.7% and 50.7% of patients in INT-1 risk group. In our cohort, prevalence of anemia and thrombocytopenia was 78.8% and 50%, respectively for INT-1 group. The prevalence of real-life ruxolitinib-related anemia may be higher than expected. Nevertheless, anemia infrequently leads to drug discontinuation.
We observed herpes zoster (N=2, 3.5%) and HBV reactivation (N=1, 1.7%) with ruxolitinib treatment who did not use prophylactic medications. An increased risk of opportunistic infections has been reported due to impaired dendritic cell functions with ruxolitinib therapy [18]. In our cohort, two patients with herpes zoster reactivation were in the INT-1 risk group. Two out of the 51 patients (∼3.9%) in the study by Palandri et al. [16] had herpes zoster infection who had no past medical history of herpes zoster reactivation. However, they administered acyclovir prophylaxis to patients with a history of herpes zoster. No cases of HBV reactivation were observed in that study. A 70-year-old male who had HBV reactivation under ruxolitinib was treated successfully with antivirals in our cohort. Hepatitis B test results were as follows at the beginning of ruxolitinib treatment: hepatitis B surface antigen (HBsAg)-negative, hepatitis core antibody (anti-HbcAg) positive, and anti-HBs negative. We strongly recommend screening for hepatitis B surface antigens, core antigens, and antibodies. Patients with isolated anti-HBc IgG positivity should also be considered for antiviral prophylaxis of hepatitis B. Since the frequency of herpes zoster reactivation is less than 10%, it is not rational to recommend antiviral prophylaxis to all patients. However, it may be prudent to administer prophylaxis for patients who have anti-varicella zoster antibodies.
Three patients had non-hematologic malignancies, including pancreatic adenocarcinoma, squamous cell carcinoma of the skin, and metastatic neuroendocrine tumor. These cancers were observed during the 1st, 2nd and 48th months of ruxolitinib treatment, respectively. All patients had a history of treatment with hydroxyurea. The patient with pancreatic cancer died soon after the diagnosis. The other patients were still alive and received anti-neoplastic treatment. Considering the timing of the cancers, there is probably no causal relationship between ruxolitinib and non-hematologic cancers. In a South Korean study, Hong et al. [19] found that the risk of developing secondary solid tumors was 2-fold higher in patients with MPN than in the normal population. Eighty (13.5%) out of 594 patients with PMF (median follow-up 31.8 mo) had secondary malignancies, and among the 85 patients with secondary myelofibrosis (median follow-up 10.3 mo) 4-year cumulative incidence rate of malignancy was 10.3%. These findings were not associated with ruxolitinib use.
The COMFORT-II trial, which was quite similar to the current study in terms of follow-up time (2.6 vs. ∼2 years, respectively) and spleen response (53.4% vs. 56.6%, respectively), reported a very high discontinuation rate in the ruxolitinib arm (73.3%). The discontinuation rate was even higher in patients who crossed over from the best available treatment arm (75.6%) [17]. However, 73.7% of patients in our cohort were still receiving ruxolitinib treatment, with a median follow-up of 22 months. As aforementioned, our cohort was younger than that of previous studies. As stem cells become older, their renewal capability decreases. The reduced hematopoietic reserves cannot properly manage drug-related hematologic toxicity. In this real-life data comprising relatively younger patients, most of the patients tolerated ruxolitinib well, and continued to use it at effective doses.
We calculated that the 2-year OS probability was approximately 75.1% and 84.5% in all cohorts and in the INT-1 risk group. Causes of death were disease progression, transformation to acute leukemia, and pancreatic adenocarcinoma. No deaths were attributed to COVID-19. In the COMFORT-II study, the 5 year-OS was reported as 56%. However, it was emphasized that these results may cause confusion due to crossover between treatment arms [17]. In contrast, an independent study evaluating the outcomes of INT-1 risk myelofibrosis estimated that 2-year OS was 80.1% [16]. We can conclude that an average 2-year OS under ruxolitinib is about 80 to 85% in patients with INT-1 risk MF.
Pooled 3-year analysis of the COMFORT trials reported that baseline anemia was associated with a shorter OS in both the ruxolitinib and control groups. However, ruxolitinib-related anemia had no adverse impact on OS [13]. We observed that patients who did not have baseline anemia tended to have a better OS within the first three years, but the survival curves merged thereafter. Patients with lower basal hemoglobin values had profound cytopenia, worse bone marrow function, and larger spleen sizes. Most of the deaths occurring within the first three years were due to disease progression rather than drug side effects. Therefore, baseline anemia is associated with advanced disease, and it is probable that ruxolitinib exerts favorable effects on survival with long-term use.
The retrospective design of this study is the most important limitation. In addition, analysis of OS is limited because of the short follow-up period. Adverse event profiles may not be complete because of reporting and detection biases. Symptom assessment was completed by less than half of the patients. Despite these limitations, our data were consistent with the findings of randomized controlled trials (RCTs).
In conclusion, ruxolitinib is effective and safe in patients with INT-1 and higher-risk MF.
 XML Download
XML Download