

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Silver–Russell syndrome (SRS, OMIM#180860) is a growth retardation disorder characterized by asymmetric fetal growth retardation and failure to catch up on growth postnatally. Since its first description by Silver [1] and Russell [2], additional clinical characteristics such as distinct facial features, relative macrocephaly, body asymmetry, clinodactyly of the fifth finger, or feeding problems, have been well established [3, 4]. To date, the underlying molecular mechanisms have been identified for approximately 50%–70% of SRS cases. Loss of methylation (LOM) of imprinting center 1 (IC1) on chromosome 11p15 and maternal uniparental disomy for chromosome 7 (mUPD7) account for approximately 30%–60% and 5%–10% of SRS cases, respectively [5-9]. Gain of methylation (GOM) of imprinting center 2 (IC2), and copy number variations and sequence variants in CDKN1C, IGF2, PLAG1, or HMGA2 were recently identified as (epi)genetic alterations associated with SRS [10-15]. Somatic mosaicisms have also been reported in SRS cases [16, 17].
A better understanding of the molecular basis of SRS may provide additional information on the disease course and prognosis for clinical practice and family counseling, and psychological stability associated with closure of the diagnostic process. However, pre- and postnatal growth retardation are relatively common conditions influenced by several maternal or other environmental factors, as well as genetic background; therefore, detailed phenotyping before genetic analysis for SRS is essential in clinical practice. Many clinical diagnostic criteria have been proposed for this disease [7,18-20], and the first international consensus on SRS in 2017 proposed a diagnostic algorithm, in which genetic analysis for 11p15 LOM and mUPD7 can be first considered for patients who meet four or more criteria of the Netchine-Harbison clinical scoring system (NH-CSS) [20]. However, some criteria cannot be always assessed in practice because of a lack of clinical data or a young patient age.
A previous retrospective study on SRS patients from Hong Kong evaluated the performance of three different scoring systems and compared their sensitivity and specificity [21]. In the present study, we used a threshold of three or more criteria of the NH-CSS for Korean patients with clinical suspicion of SRS to identify the practical utility and limitations of the system for the first time and performed multi-staging molecular and in-depth phenotypic analyses.
MATERIALS AND METHODS
Patients and clinical information
This study, approved by the Institutional Review Board (IRB) of Seoul National University Hospital, Seoul, Korea (IRB number 1805-156-948), was conducted between January 2018 and July 2020. Twenty-eight patients or their legal representatives provided written informed consent. Inclusion criteria were determined based on the NH-CSS [22]: (1) small for gestational age [SGA; birth weight and/or birth length ≤2 standard deviation score (SDS)], (2) postnatal growth failure (height at 24±1 months ≤−2 SDS or height ≤−2 SDS below the midparental target height), (3) relative macrocephaly at birth (head circumference at birth ≥1.5 SDS above the birth weight and or length SDS), (4) frontal bossing [forehead projecting beyond the facial plane on a side view as a toddler (1–3 years)], (5) body asymmetry [leg length discrepancy (LLD) ≥0.5 cm, arm asymmetry, or LLD <0.5 cm with at least two other asymmetrical body parts (one non-face)], (6) feeding difficulties and/or low body mass index (BMI ≤−2 SDS at 24 months) or a history of feeding tube use in infancy. We enrolled patients who met three or more of the above criteria of the NH-CSS. We further modified the conventional application of NH-CSS criteria for assessing older patients without detailed infantile or perinatal records. In particular, relative macrocephaly at birth was replaced by anthropometric values at the initial examination, if birth data were unavailable; feeding difficulty was defined as feeding tube usage until a postmenstrual age of 44 weeks; and postnatal growth failure and low BMI were also considered to be present, if the value measured nearest to 24 months of age was below 2 SDS. Patients who had other known causes for growth retardation were excluded from the study. Among the 28 patients enrolled, 15 (53.6%) were males. The median age at the first examination was 12 months (range, 0–117 months), and the median follow-up duration was 19 months (range, 1–137 months).
Physical growth data were evaluated based on Korean reference data and were expressed as SDS [22]. All data, including birth history, serial growth and developmental status, and examination results, were collected from medical records.
Genetic/epigenetic analysis
Peripheral blood samples (6 mL) were obtained from 28 patients. Genomic DNA was isolated from peripheral blood leukocytes using a Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. A two-step molecular analysis strategy was adopted. The genomic DNA was subject to methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) on chromosome 11p15 as the first diagnostic step. Bisulfite pyrosequencing (BP) analysis for IC1 and IC2 at chromosome 11p15 was then performed in all patients to validate the MS-MLPA results. BP for MEST on chromosome 7q32.2 and MEG3 on chromosome 14q32.2 was also performed in patients who did not exhibit methylation defects on IC1 and IC2 at 11p15.
MS-MLPA analysis was conducted using a SALSA MLPA kit (ME030 BWS/SRS, MRS Holland, Amsterdam, Netherlands) according to the manufacturer’s instructions. Probes were hybridized to denatured genomic DNA, and hybridized samples were divided into two portions for separate analyses: (1) direct ligation or (2) digestion with the HhaI methylation-specific restriction enzyme before ligation. After ligation, PCR was performed using fluorescence-labeled unique primers for probe sets provided in the SALSA MLPA kit, ME030 BWS/SRS, including IC1 and IC2. The former analysis identifies changes in the copy number of CDKN1C, KCNQ1, KCNQ1OT1, IGF2, and H19, and the latter analysis determines the methylation status of IC1 and IC2 on 11p15. Amplified products were separated on an ABI3130xl Genetic Analyzer (Applied Biosystems, CA, USA) and analyzed using GeneMarker v.1.9 software (SoftGenetics, Pittsburgh, PA, USA).
BP was performed for IC1 and IC2 on 11p15, MEST on 7q32.2, and MEG3 on 14q32.2 using internally designed targeted assays covering 3–7 consecutive CpG sites for each locus (Table 1). Sodium bisulfite-modified genomic DNA was amplified using the Hot-Start Taq master mix (Qiagen, Hilden, Germany). Regions of interest were amplified using PCR, and pyrosequencing was carried out using a PyroMark Q24 pyrosequencer (Qiagen) according to the manufacturer’s protocol. Output data were analyzed using PyroMark Q24 1.0.10 Software (Qiagen), which calculates the methylation levels [%mC=mC/(mC+C)] for each CpG site, allowing for quantitative comparisons. Altered DNA methylation levels (%mC) at IC1 or IC2, MEST, and MEG3 were calculated based on the average DNA methylation level detected in 20 samples from age- and sex-matched healthy Korean children (as controls), who agreed with the present study and written consent.
Statistical analysis
Statistical analyses were performed to compare clinical features between mutation positive and negative groups and determine the factors that favor such (epi)genetic defects. The Student’s t-test was employed for continuous variables and Pearson’s chi-square test for categorical ones using the IBM SPSS Statistics 22 software suite (IBM Corp., Armonk, NY, USA). The value of statistical significance was set at P<0.05. Statistical processing of BP result for the control group was conducted using the Microsoft Office Excel software. Reference values are presented as the mean±2SD, and methylation levels outside the reference values were considered as GOM or LOM.
RESULTS
Phenotypic characteristics
According to the NH-CSS, 13 patients (46.4%) met three diagnostic criteria while eight (28.7%), five (17.9%), and two (7.1%) patients met four, five, and six criteria, respectively. All except for two (92.9%) patients had low birth weight (≤−2 SDS). Postnatal growth was retarded in 25 patients (89.3%), and 18 (64.3%) patients showed relative macrocephaly. A frontal bossing and clinodactyly of the fifth finger were noted in 16 (57.1%) and 14 (50.0%) patients, respectively. LLD was noted in 13 (46.4%) patients.
Recombinant human growth hormone (rhGH) therapy (35–70 μg/kg daily) was started in 10 patients at a median age of 4.8 years (range, 2.8–9.8 years). Six patients continued the treatment until the last follow-up, with an average treatment duration of 4.0 years (range, 1.3–8.3 years), while four patients discontinued treatment after around 5.0 years (range, 2.5–11.5 years) of treatment initiation. After rhGH treatment, five patients showed catch up in height over+1.5 SDS.
Molecular analysis and diagnostic yield
Epigenetic defects were confirmed in 17 of the 28 patients, for an overall diagnostic yield of 60.7% (Table 2). MS-MLPA identified LOM in IC1 (IC1-LOM) in 11 patients (39.3%). None of the patients had a copy number variation at 11p15. BP for 11p15 detected methylation defects in the 11 patients for whom a diagnosis was obtained using MS-MLPA and in three patients (10.7%) who did not show abnormal results on MS-MLPA (Patients 7–9). Patient 7 was SGA (−2.69 SDS) and failed to catch up on growth; he showed no definite macrocephaly or distinctive facial characteristics but had a definite LLD (NH-CSS score of 3). Patients 8 and 9 were also SGA, exhibited postnatal growth retardation, and had distinctive facial characteristics suggestive of SRS along with relative macrocephaly. The BP results for 7q32 and 14q32.2 led mUPD7 diagnosis in 3 (10.7%) patients; however, none of these patients had methylation defects on 14q32.2.
Six of the seven patients (85.7%) who met five or more criteria of the NH-CSS were confirmed to have methylation defects (IC1-LOM in five patients and mUPD7 in one patient). Molecular confirmation was achieved for four out of the eight (50.0%) patients with four NH-CSS criteria, and for seven out of the 13 patients (53.8%) with three NH-CSS criteria, which did not represent a significant difference in diagnostic yield.
Comparison of the genetic and phenotypic spectrum
The average gestational age and body weight were similar between patients with and without (epi)genetic variants (Table 3). The frequencies of each NH-CSS criterion were comparable between the two groups, except for body asymmetry, which was more frequent in the patients with variants (P=0.024). Additionally, relative macrocephaly was noted more frequently in the patients with (epi)genetic variants (P=0.097).
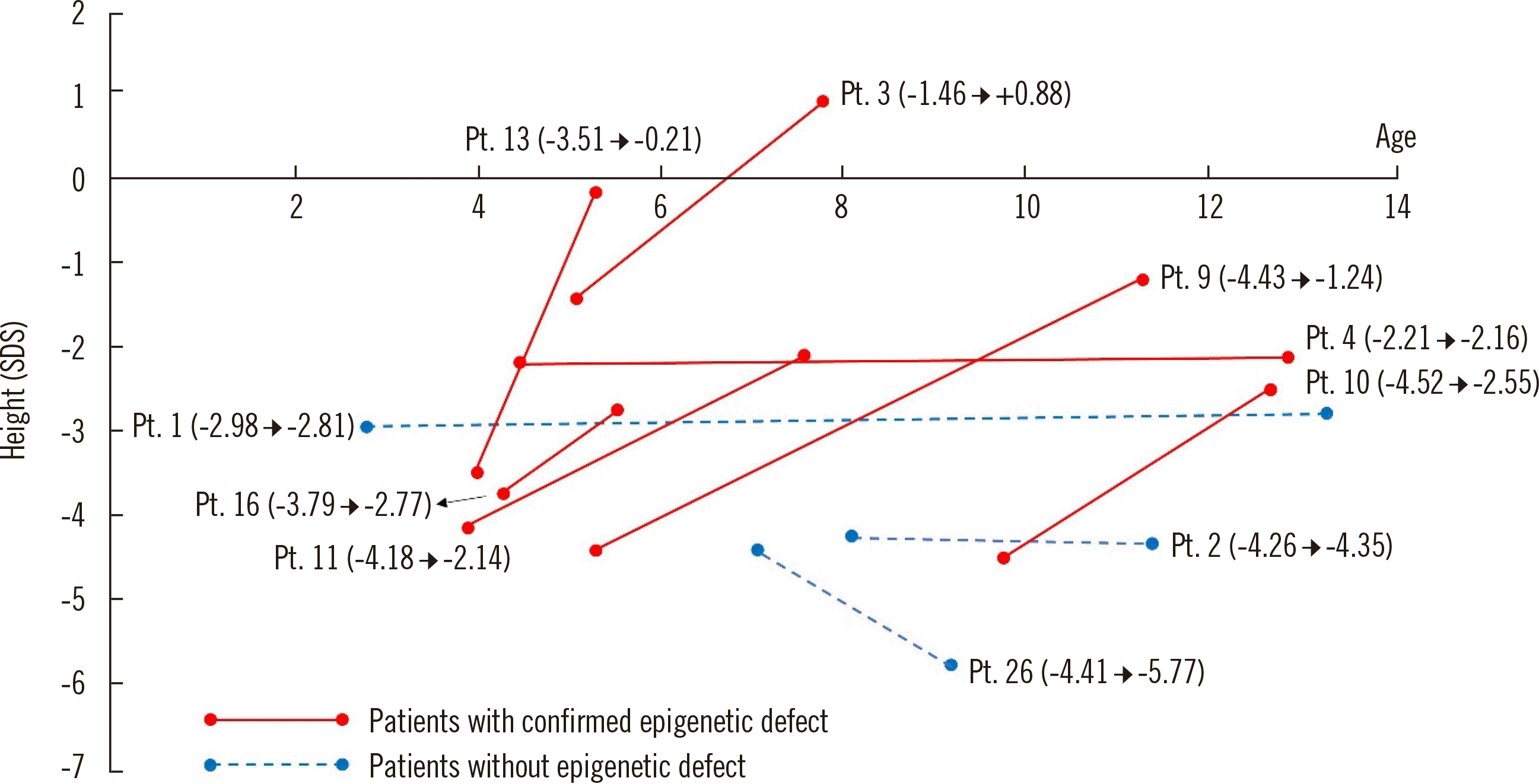
The rhGH treatment response was different according to variant status (Fig. 1). However, the age at treatment and doses of rhGH administered were not different between the patients with or without epigenetic defects. Six of the seven patients who carried a methylation defect (IC1-LOM in four patients and mUPD7 in two patients) showed catch up in height (average +2.0 SDS, range +0.05 to +3.30 SDS) during the treatment period (median duration, 50 months; range, 5–100 months). In contrast, the rhGH treatment response was worse in the remaining three patients who did not have a methylation defect.
DISCUSSION
In this study, the modified application of the NH-CSS criteria exhibited powerful predictive value for the screening of patients with SRS, despite its limitations in practice. The overall diagnostic yield was 60.7%, which was comparable to the results of other large-cohort studies [20, 24, 25]. In particular, a similar diagnostic yield was obtained for patients who met three or four criteria of the NH-CSS (53.8% vs 50.0%). In detail, seven out of the 13 patients who met three NH-CSS criteria were confirmed to have a methylation defect, and all of them had limb asymmetry and/or relative macrocephaly. Therefore, these results suggest SRS diagnosis and the necessity for active molecular analysis in borderline patients assessed using the NH-CSS. Nevertheless, other clinical characteristics, including frontal bossing, a triangular face, and feeding difficulties, remain important criteria for the screening of SRS.
The additional diagnostic yield of BP is well established [25, 26]. We conducted a two-step diagnostic test and obtained additional genetic confirmation of IC1-LOM in three patients (10.7%; Patients 7, 8, and 9). These patients were not borderline patients but rather showed characteristics leading to a high suspicion of SRS. However, their methylation levels from MS-MLPA were located within the 95% confidence interval of the reference values and could not be classified as abnormal, even though the probe ratios slightly deviated from the center of the ratio chart. The limitations of MS-MLPA are well known, and clinicians should bear in mind the possibility of false-negative results, especially in cases of high suspicion for SRS [24, 26]. In contrast to MS-MLPA, BP analysis provides quantitative values for methylation abnormalities and increases the diagnostic yield, particularly in cases with mild and marginal methylation abnormalities [24]. Therefore, BP should be considered in patients for whom a diagnosis of SRS is highly suspected, even if they showed negative results in MS-MLPA.
Three patients with mUPD7 (Patients 10, 11, and 21) were also diagnosed as having SRS using BP. We did not find any differences in auxological parameters between the IC1-LOM and mUPD7 subgroups. According to previous studies, patients with mUPD7 tend to have more neurocognitive problems, including global developmental delay or learning disability, whereas classic SRS phenotypes are noted more frequently in patients with IC1-LOM [20, 27]. In the present cohort, none of the patients with mUPD7 had body asymmetry, even though they had facial characteristics and other clinical findings typical of SRS. Moreover, none of these patients exhibited neurocognitive problems. The degree of methylation abnormality detected by BP was also not clearly correlated with the NH-CSS score or phenotypic severity in our cohort. These results suggest that patients with SRS with different genotypes are on a “phenotype continuum” rather than in distinguishable subgroups.
The phenotypes themselves did not provide clues regarding which genetic analysis should be conducted first. However, the phenotype-genotype correlation may be more informative once the remaining unknown (epi)genetic background, which accounts for 30%–40% of the total number of SRS cases, is identified. Indeed, many studies on rare genetic alterations are ongoing; however, we could not perform further scheduled molecular analysis due to limited resources. For patients negative for epigenetic alterations in our cohort, chromosomal microarray would be recommended first, especially for the ones with additional anomalies or developmental delay, while next generation sequencing could be the next step [28, 29]. Among the 11 patients without an (epi)genetic variant, additional analyses were performed using chromosomal microarray or whole-exome sequencing for five patients, and a diagnosis of 3M syndrome was ultimately made for one patient with compound heterozygous variants of CUL7 in the clinic. Six patients were lost to follow-up.
The effectiveness of rhGH treatment in patients with SRS has been well demonstrated in many studies, including clinical trials and long-term observational studies [30–33]. However, most of these studies included patients diagnosed as having clinical SRS without (epi)genetic confirmation. Binder, et al. [32] also classified patients according to their epigenotype (11p15-positive, mUPD7-positive, and negative) and did not find significant differences in treatment response among the three groups. Our cohort was mainly generated for diagnostic confirmation using epigenetic analyses; therefore, only a small number of patients received rhGH treatment. The genetic background of the patients with a negative result from (epi)genetic analysis is unknown and can be rather heterogeneous compared with that of the patients with epigenetic alterations. Therefore, we cannot confirm the causes of the different responsiveness to rhGH treatment between the patients with and without (epi)genetic variants. However, for SRS patients with a known methylation defect, rhGH treatment should be offered to the parents as a treatment option.
The present study has some limitations. The number of study participants was not enough to obtain strong clinical significance from statistical analysis. We suggest the clinical utility of the NH-CSS scoring system but applied it in a modified manner as data for some patients was missing from retrospective records.
In conclusion, SRS is a clinical entity that requires detailed phenotyping in clinical practice. Genetic and epigenetic evaluations might be helpful for establishment of a molecular diagnosis, even in patients not meeting four or more of the NH-CSS criteria, especially if they have limb asymmetry or relative macrocephaly. BP analysis can be added to MS-MLPA or performed as a first-tier test in cases of high clinical suspicion. Such molecular confirmation could facilitate providing appropriate and timely medical support.
 XML Download
XML Download