

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Hepatocellular carcinoma (HCC) is the second-most-common cause of cancer-related deaths worldwide [1]. In Korea, the mortality rate of HCC was 21.5 per 100,000 population in 2016, and HCC ranked as the second-leading cause of cancer-related deaths [2]. The major risk factors of HCC include chronic hepatitis B virus (HBV) and hepatitis C virus infection, alcohol abuse, liver cirrhosis, and exposure to aflatoxin B1 [3]. Alpha-fetoprotein (AFP) is the most widely used biomarker for early detection of HCC. However, AFP has poor reliability and low sensitivity for early-stage HCC detection [4]. The lack of an early diagnostic marker for HCC has posed a major challenge to curative treatments, including liver resection and liver transplantation. In addition, early detection of recurrence during monitoring after curative therapy is associated with improved survival in HCC patients [5]. Thus, for early detection of HCC as well as for recurrence monitoring after curative surgical resection, an accurate and non-invasive HCC biomarker is required [6].
Circulating cell-free DNA (cfDNA) has potential as a noninvasive biomarker for detecting and monitoring tumor cells. In addition, cfDNA has prognostic value and may be useful in strategies to select patients eligible for targeted therapy [7, 8]. Several studies have demonstrated significantly higher cfDNA levels in sera and plasma of patients with both early and advanced HCC [9, 10]. Recent studies have explored HCC-related genomic alterations and have identified frequent gene variants, including those in the TERT promoter, TP53, and CTNNB1 (beta-catenin), in both early and advanced HCC samples [11, 12]. However, the analysis of cfDNA is challenging because of its short half-life and low concentration in the plasma; it is even more challenging to detect tumor-derived cfDNA, which accounts for a very small fraction of the total cfDNA [13]. Therefore, various methods have been applied for cfDNA analysis, including quantitative PCR, digital PCR, BEAMing, and next-generation sequencing (NGS) [14]. While all these methods enable sensitive detection of target genes, only NGS facilitates the parallel detection of a broad range of variants of multiple gene targets [15]. This is especially important for HCC, as most somatic variants in HCC are not located in hotspots, and this is relevant even for variants in HCC driver genes [16, 17].
In this study, we aimed to assess pathogenic variants of HCC driver genes in cfDNA from advanced HCC patients who had not undergone systemic therapy. We used a customized targeted NGS panel that incorporates unique molecular identifiers (UMIs) to reduce PCR-based NGS errors and to distinguish reads amplified from the same original DNA molecule (on the basis of identical UMIs).
MATERIALS AND METHODS
Participants
Twenty patients, including 17 men and 3 women with a median age of 60 years (range: 47–79 years), admitted at Seoul St. Mary’s Hospital, Seoul, Korea, between June 2018 and May 2019 were enrolled in this study. All patients were diagnosed as having HCC according to the guidelines from the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver [18]. Cirrhosis was present in all 20 (100%) patients, with viral hepatitis B being the main etiology for the underlying liver disease in 15/20 (75%) patients. Most patients had multiple nodules (17/20, 85%), and macrovascular invasion and metastasis were present in 9/20 (45%) and 14/20 (70%) patients, respectively (Table 1). All participants, including three healthy adults recruited as healthy controls, provided written informed consent. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board/Ethics Committee of Seoul St. Mary’s Hospital (IRB No. K18TESI0295).
Sample collection and cfDNA extraction
Peripheral blood samples (10 mL) were drawn in ethylenediaminetetraacetic acid (EDTA)-containing tubes, and plasma was separated within one hr of collection in two centrifugation steps: 2,000 × g at 4°C for 10 minutes, followed by 16,000 × g at 4°C for 10 minutes [19]. Plasma samples were immediately aliquoted and stored at –80°C for up to nine months.
Circulating cfDNA was isolated from 4 mL of plasma using the MagMAX Cell-Free DNA Isolation Kit (Applied Biosystems, Waltham, MA, USA) and the KingFisher Duo Prime Magnetic Particle Processor (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. The size of the purified plasma DNA was estimated using a 2,100 Bioanalyzer System (Agilent Technologies, Santa Clara, CA, USA), and its concentration was determined using a Qubit fluorometer (Thermo Fisher Scientific) in combination with a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Seraseq ctDNA Reference Material v.2 (SeraCare Life Sciences, Milford, MA, USA) was used to validate the limit of detection. The reference material consisted of 40 cancer-relevant somatic variants spiked into a background of wild-type DNA (purified from a reference cell line, GM24385) at defined variant allele frequencies (VAFs) of 2%, 1%, 0.5%, 0.25%, 0.125%, and 0% [20]. Seraseq ctDNA was extracted and analyzed in duplicate. Methods used for library preparation and sequencing were the same as those used for the participant samples.
Targeted NGS
We designed a custom 88-amplicon panel (mean read length: 107 bp) targeting three HCC driver genes, namely TP53, CTNNB1, and TERT, using the Ion AmpliSeq Designer (Thermo Fisher Scientific) and the “white glove” design option, a program that facilitates additional customization to design amplicons for difficult regions. Two primer pools were designed to interrogate the entire coding regions of TP53 and CTNNB1, including 5 bp of the 5’ and 3’ intronic sequences of each exon, and TERT promoter variants (primer sequences can be provided on request). The target design rate was 100% for TP53, CTNNB1, and for TERT, totaling 3.92 kb. The Ion AmpliSeq HD panel enabled molecular tagging of each DNA input molecule using UMIs.
We used the optimal amount of input cfDNA (20 ng in 8.3 µL) recommended by the manufacturer to generate libraries using an Ion AmpliSeq HD library kit (Thermo Fisher Scientific) and the Custom Ion AmpliSeq HD panel. Library quantification was performed using the TapeStation 2200 High Sensitivity D1000 Kit (Agilent Technologies). Clonal amplification of the libraries was performed by emulsion PCR on an Ion Chef System using an Ion 540 Kit-Chef (Thermo Fisher Scientific). Template-positive ion sphere particles were enriched, loaded on an Ion 540 Chip, and sequenced using an Ion S5 XL Sequencer (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Sequence data were processed for primary and secondary analyses, using standard Ion Torrent Suite Software (Thermo Fisher Scientific) running on the Torrent Server (Thermo Fisher Scientific). Raw signal data were analyzed using Torrent Suite v. 5.10.1 (Thermo Fisher Scientific) and Ion Reporter (Thermo Fisher Scientific). The pipeline included signal processing, base calling, quality score assignment, adapter trimming, PCR duplicate removal, read alignment, mapping quality control, coverage analysis, and variant calling. The sequenced reads were aligned against the hg19 reference genome (Genome Reference Consortium GRCh37). Sequence variants were identified using the Ion Reporter software v. 5.10 (Thermo Fisher Scientific) and Ion AmpliSeq HD Workflow template for Liquid Biopsy-w2.1-DNA-Single Sample, and the coverage of each amplicon was determined using the Coverage Analysis Plugin Software v. 5.10.0 (Thermo Fisher Scientific). The application of UMIs enabled the grouping of reads into molecular families. Random errors generated during library construction and the sequencing process were removed automatically. At least three independent molecular families were required to identify and call a variant.
Statistical analysis
Categorical clinical variables between patients with and without pathogenic or likely pathogenic variants identified in the cfDNA were compared using Fisher’s exact test. Observed and designed VAFs were compared using Spearman’s rank correlation and Passing–Bablok regression. Statistical analyses were performed using MedCalc v. 17.2 (MedCalc Software, Ostend, Belgium). P < 0.05 was considered statistically significant.
RESULTS
cfDNA amount and summary of the sequencing metrics
The cfDNA output of each sample and the sequencing metrics are presented in Table 2. The median concentration of plasma cfDNA from all HCC patients was 4.1 ng/mL (range: 0.8-15.3 ng/mL). The library concentrations were 2,050–14,500 pM. The median sequencing coverage was 62,694 (range: 44,765–81,672), and all 20 samples had a median read coverage greater than 25,000, which is the median read coverage across targets specified by the manufacturer to ensure a 0.1% limit of detection.
Validation of the limit of detection using reference material
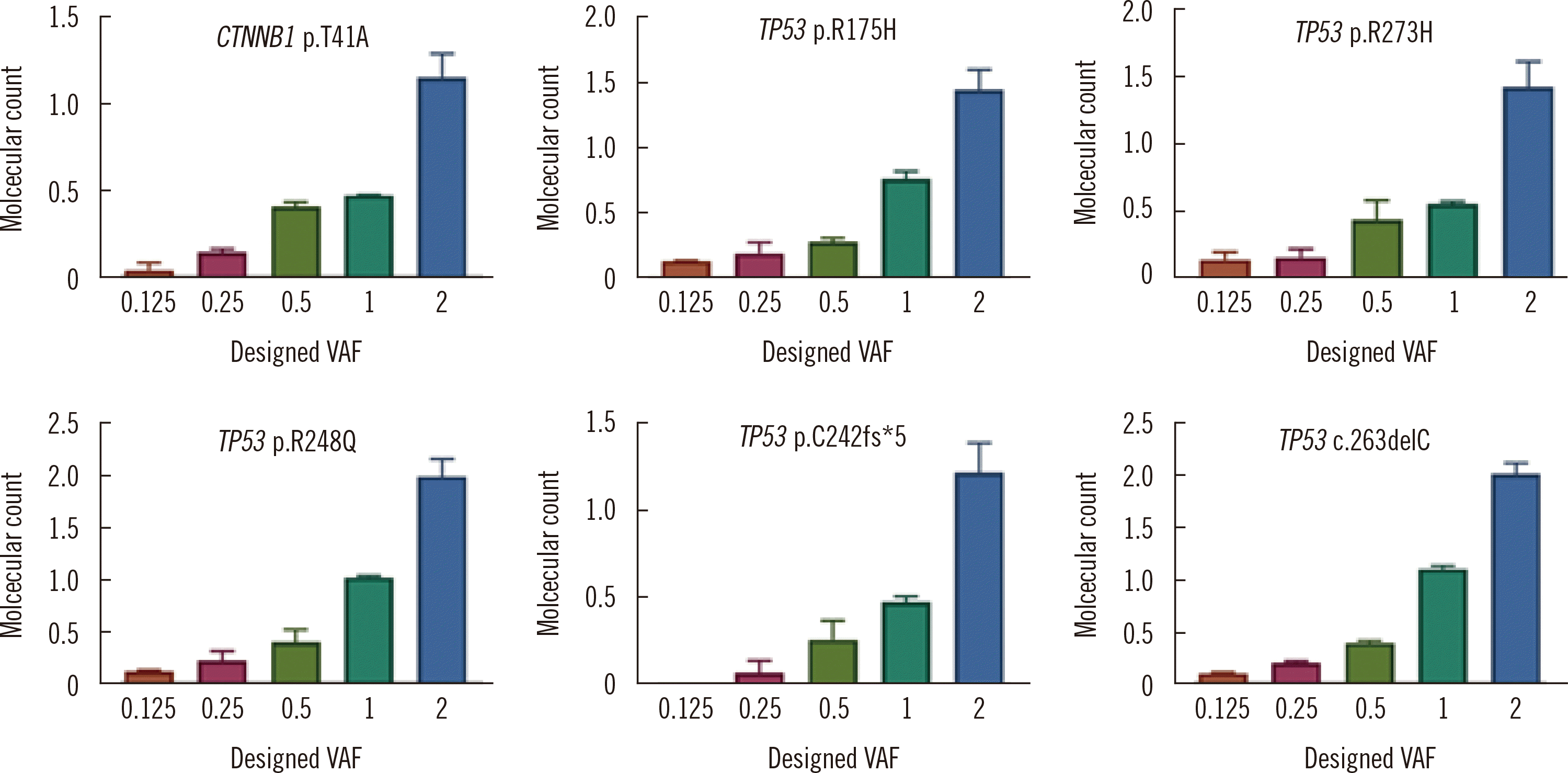
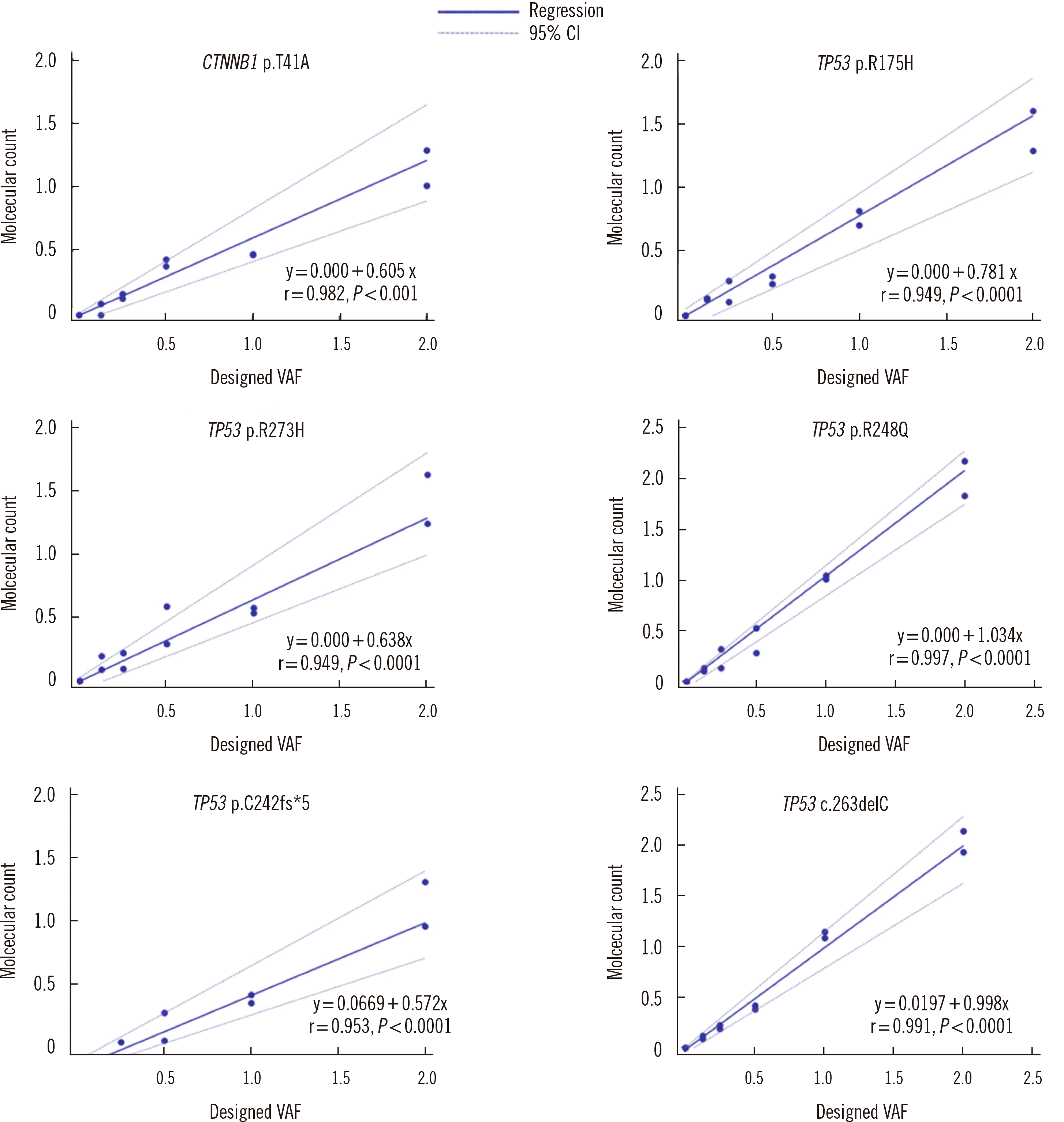
The results of the analysis of the reference materials for six variants (one in CTNNB1 and five in TP53) included in our custom panel design are shown in Fig. 1. Of the six variants, only TP53 p.C242fs*5 was detected at 0.25%; all other variants were detected at 0.125%. The Spearman’s rank correlation coefficient for each variant ranged from 0.95 to 0.99, indicating a strong correlation between the observed and designed VAFs. Passing–Bablok regression results for comparisons of the observed and designed VAFs for each of the six variants are shown Fig 2. The slope of the linear regression line for each variant ranged from 0.57 to 1.03, and for the TP53 p.R248Q and TP53 p.C242fs*5 variants, the 95% confidence interval of the slope contained 1.0.
Low-frequency variant detection in three HCC driver genes
Targeted NGS using an in-house panel of three HCC driver genes identified at least one pathogenic variant in the plasma cfDNA of 13/20 patients (65%). These included 16 variants of TP53 and nine variants of CTNNB1. The TP53 and CTNNB1 variants had low-level allele frequencies with median values of 0.17% (range: 0.06%–6.99%) and 0.07% (range: 0.05%–0.96%), respectively. However, the molecular coverages of the TP53 and CTNNB1 variants detected in cfDNA were sufficient, with median values of 5,543 (range: 2,317–9,088) and 7,568 (range: 2,400–9,633), respectively. Of note, no pathogenic variants were identified in the three healthy control samples.
Of the 16 pathogenic variants of TP53, 12 (75%) were missense, 13 (81%) were located in the DNA-binding domain in exons 4–8 of TP53, and two (13%) were located at codon 245, which is one of the major hotspots of somatic TP53 pathogenic variants. Of the nine variants of CTNNB1, four were located at known hotspots [18-21], two (S33Y, G34Y) were located within the β-TrCP binding domain (D32-S37), one (S45F) involved S45, an amino acid residue involved in the phosphorylation/degradation of β-catenin, and one was located in the armadillo repeat six domain (N387). Probably due to the small number of patients included in this study, we did not find a statistically significant association between clinical characteristics and the presence of TP53 and/or CTNNB1 cfDNA variants (Table 3).
DISCUSSION
In the present study, we identified 16 pathogenic variants of TP53 and nine pathogenic variants of CTNNB1 from the cfDNA of patients with advanced HCC. In both TP53 and CTNNB1, relations between somatic variants and prognosis in HCC were identified [7, 21]. The presence of TP53 pathogenic variants in HCC was significantly associated with shorter survival and disease-free survival, and the R249C variant, one of the most common TP53 pathogenic variants in HCC, was a better indicator for poor prognosis than the TP53 pathogenic and likely pathogenic variants combined [7]. Recent HCC variant screening studies have shown significant progress at identifying potentially targetable pathogenic variants [8, 22]. Therefore, in addition to early detection and monitoring of HCC, cfDNA can be useful for prognostication and in strategies to select patients eligible for targeted therapy.
Two recent studies investigated the molecular landscape of cfDNA in HCC patients [22, 23] (Table 4). In the European study, 29 pathogenic or likely pathogenic variants in eight genes were detected in 18/51 (35%) patients. The median read depth was 486, and the median VAF was 0.12. In the study on 206 HCC patients from the USA, alterations, including amplifications, synonymous alterations, and variants of undetermined significance, in addition to pathogenic or likely pathogenic variants, were detected in 181/206 (87.8%) patients. The median VAF was 0.49%, and TP53 was the most commonly altered gene.
Although these were larger scale studies than our study, to the best of our knowledge, our study is the first to use a customized cfDNA NGS panel targeting the major HCC driver genes, incorporating molecular barcoding. Using an in-house customized panel targeting three HCC driver genes implemented on a robust and standardized NGS platform, we detected at least one pathogenic variant in plasma cfDNA among 68% of the patients analyzed (13/19). These included 16 variants of TP53 and nine variants of CTNNB1, and 13% of TP53 and 44% of CTNNB1 variants were located in reported hotspot regions. Of note, a possible reason that we did not detect the TERT promoter variant may be that it is more frequently seen in early-stage HCC [11], and this study did not include early-stage HCC patients. Importantly, all cfDNA variants identified in this study were present at very low frequencies, with median values of 0.17% and 0.07% for TP53 and CTNNB1 variants, respectively. This low frequency supports the use of a robust cfDNA NGS panel that incorporates molecular barcodes for achieving optimal sensitivity. Incorporation of molecular barcoding has significantly reduced false-positive errors associated with amplicon-based NGS methods and thereby allows for a sensitivity of variant detection down to -0.001% [24]. We validated the detection limit of our customized cfDNA NGS panel using commercially available reference material.
We acknowledge some limitations of our study. First, somatic variant profiles from synchronously collected tumor biopsies were not available for concordant analysis. However, a previous study indicated that, even without prior knowledge of the variant repertoire in an HCC biopsy, high-depth sequence analysis of plasma cfDNA can represent somatic variants in an HCC biopsy in a significant proportion of therapy-naive HCC patients [25]. Second, as a pilot study, our study enrolled small number of patients. However, we found strong evidence that plasma cfDNA analysis using NGS can reliably detect pathogenic variants in HCC driver genes in HCC patients. Furthermore, as a proof-of-concept study, correlations between detected cfDNA variants and clinical characteristics were not the main focus of this study. These should be assessed in a larger patient cohort in future studies. Lastly, we did not confirm the very-low-frequency variants using other methods, such as digital droplet PCR.
In conclusion, by using targeted cfDNA NGS, we achieved a very high coverage of the entire coding regions of three major HCC driver genes, allowing the detection of low-frequency variants in a large number of unique molecular families. cfDNA could be used as a reliable biomarker to identify somatic variants in HCC, and our results support the utility of cfDNA analysis in a larger cohort of HCC patients.
 XML Download
XML Download