

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Alzheimer’s disease (AD) is pathologically characterized by the involvement of extracellular deposition of plaques comprised of Aβ peptide, and intracellular neurofibrillary tangles (NFT), which lead to neurological damages and cognition/memory deficits [1]. The amyloid hypothesis has been considered as the main pathogenesis of AD for two decades. Due to this reason, some of the earliest proposed therapeutic strategies have targeted the elimination of Aβ peptides and deposits.
Cleavage of APP by BACE1 (β-site APP cleaving enzyme) generates a membrane-bound soluble C-terminal fragment, and subsequent cleavage by γ-secretase further produces Aβ1-40 and Aβ1-42 in brain [2]. Otherwise, the activation of α-secretase cleaves APP in its extracellular domain and releases a large soluble N-terminal ectodomain, sAPPα, which reduces Aβ peptide production [3]. α-Cleavage is of pharmacological interest because secreted sAPPα was approved neuroprotective in AD models [4].
GSK-3β is activated by Aβ peptides [5] and increased GSK-3β activity accounts for the abnormal tau hyperphosphorylation, and the accumulation of aberrantly phosphorylated tau, which underlies tau-mediated neurodegeneration [6,7]. Inhibition of GSK-3β reduces the production of Aβ peptides in amyloid plaques [8]. Interestingly, cilostazol increased GSK-3β phosphorylation at Ser 9 (inactive form) and β-catenin phosphorylation at Ser675 in cytosol and nuclei and thereby, suppressed Aβ1-40-induced neurotoxicity and enhanced neurite elongation [9]. Evidence has accumulated that alterations in nerve-growth factor (NGF)/TrkA signaling cause selective degeneration of cholinergic basal forebrain neurons in AD [10,11]. Based on these evidences, cholinergic hypothesis was postulated that cholinotropic agents can be a logical choice for preventative therapy in AD [12,13].
In recent years, the role of sirtuins in AD has become increasingly apparent [14-16]. Julien et al. [17] have emphasized a close relationship between decrease in SIRT1 levels and tau accumulation in AD and Aβ accumulation in the brains of patients with AD. It has been demonstrated that SIRT1 activates transcriptions of gene encoding α-secretase/ADAM10 (a disintegrin and metalloproteinase 10) via the deacetylation of retinoic acid receptor-β (RARβ) [15,18]. Cilostazol was reported to suppress accumulations of full-length APP and Aβ by activating ADAM10 via upregulating SIRT1-coupled RARβ activation [19]. In line with these, it has been reported increased CREB (cAMP-response element binding protein) phosphorylation by cilostazol is associated with elevated levels of phosphorylated CK2, Akt, and Bcl-2 with reduced caspase-3 activity in the penumbral region of focal ischemic rat brain [20,21]. Park et al. [22] found cilostazol reduced intracellular Aβ levels and phosphorylated tau levels in activated neuro2A (N2a) Swe cells, and significantly improved spatial learning and memory in the Aβ-injected mice.
On the other hand, aripiprazole as an atypical antipsychotic with partial agonistic activity, acts on dopamine D2 and 5-HT1A receptors, and has a potent partial agonistic effect on 5-HT1A receptors and an antagonistic effect on 5-HT2A receptors [23]. Aripiprazole was licensed by the Food and Drug Administration (FDA) and European Medicine Agency (EMA) for treatment of schizophrenia [24] and major depression [25]. Declining of cognitive function in AD patients is prevalently accompanied by psychotic symptoms and behavior [26,27]. De Deyn et al. [28] reported that aripiprazole-treatment showed significant improvements in psychiatric rating scale vs. placebo treatment in AD patients with psychosis. Recently, Park et al. [29] reported aripiprazole ameliorates Aβ1-42-evoked toxicity associated with increased neurite outgrowth and viability in neuronal cells.
At present time, only five prescription drugs are approved by the US FDA for the symptomatic treatment of AD [30]. These are: (i) donepezil, (ii) galantamine, (iii) memantine (a non-competitive N-methyl-d-aspartate [NMDA] receptor antagonist.), (iv) rivastigmine, and (iv) donepezil and memantine. AD is associated with neurodegenerative changes associated with depletion of nAChRs and loss of cholinergic neurons, thereby resulting in progressive cognitive and functional deficits clinically [31,32]. Donepezil (R,S-1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methylpiperidine hydrochloride) (E2020), a potent and reversible acetylcholinesterase (AChE) inhibitor, has been reported to show moderate improvement of cognitive function with minimal side effects [33] and early prescribed for treatment of cognitive dysfunction in AD [34].
Since the pathogenesis of AD is known to be multifactorial, and current single drug-based strategies have been proven to be inadequate, in this study we proposed an alternative therapeutic approach: a multitarget-directed drug combination including cilostazol plus aripiprazole with donepezil add-on for modulation of Aβ accumulation, phosphorylated-tau, GSK-3β, α-secretase/ADAM10, and SIRT1 levels in N2a Swe cells and for revitalization of acetylcholine (ACh) release and choline acetyltransferase (CHAT) expression.
METHODS
Experimental groups in vitro study
Group 1: N2a cells [control].
Group 2: N2a Swe cells activated by culturing in 1% FBS-containing medium for 24 h [vehicle].
Group 3: After pretreatment with cilostazol (3 μM) /aripiprazole (3 μM), N2a Swe cells were activated [CA group].
Group 4: After pretreatment with cilostazol/aripiprazole + donepezil (DNP; 3 μM), N2a Swe cells were activated [CAD group].
Group 5: N2a Swe cells pretreated with donepezil (3 μM) and then activated [DNP group].
Cell culture
Mouse neuroblastoma N2a and N2a Swe mutant cells, were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) at 37°C in a 5% CO2/95% air. For evoking endogenous Aβ overproduction, the culture medium was switched from medium containing 10% FBS to medium with 1% FBS, and then cultured for 3, 12, 24, or 48 h as described by Lee et al. [35]. When drug treatment was required, cells were previously treated for 3 h and then exposed to medium containing 1% FBS. HT22 cells were also maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS.
Western blotting
Following stimulation with drugs or inhibitors, N2a Swe mutant cells were scraped and lysed in lysis buffer [35]. After centrifugation at 13,000 rpm for 7 min, 30 μg of total protein was loaded onto 10% SDS-polyacrylamide gels. Subsequently, separated proteins were transferred to nitrocellulose membranes. Membrane was blocked with 5% skim milk (at 4°C overnight) and incubated with antibodies against anti-Aβ (6E10) and SIRT1 (Covance, Emeryville, CA, USA) (1:500 dilution), GSK-3β, GSK-3β phosphorylated at Ser9 (GSK-3β P-Ser9), anti-p-Tau (p-Ser396; Sigma-Aldrich, St. Louis, MO, USA), and anti-ac-Tau (Acetyl lys174; Signalway Antibody, College Park, MD, USA). Antibody against P300, ADAM10 and GSK-3β phosphorylated at Tyr216 (1:500 dilution) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and anti-p-CREB Ser133 was from Cell Signaling Technology, Danvers, MA, USA. Membranes were probed with an anti-β-actin antibody (MP Biomedicals, LLC, Solon, OH, USA) as an internal control. DMSO was used as vehicle (< 0.1% v/v of final volume).
Assay of α-secretase activity
As described by Lee et al. [19], N2a Swe cells were cultured in DMEM supplemented with 10% FBS. α-Secretase activities were analyzed in cultured cell lysates using a kit (No. AS-72085) Fluorimetric; ANASpec, Fremont, CA, USA). Cell membranes were homogenized in assay buffer containing 0.1% (v/v) Triton X-100. After addition 50 μl of stop solution to each well, fluorescence intensities were determined at excitation and emission wavelengths 490 and 520 nm, respectively. The assay was assayed three times in duplicate.
Measurement of cholinergic function markers
CHAT expression: Cultured N2a and N2a Swe cells were homogenized in 9 volumes of cold saline and centrifuged at 3,000× g to obtain supernatants, and these were diluted with a buffer solution. Protein concentrations of the supernatants were measured by Coomassie blue method. Anti-CHAT (1:500 dilution) was from Millipore (Temecula, CA, USA). CHAT expressions were assessed by Western blotting.
Acetylcholine assay: N2a Swe cells were destroyed by repeated freezing and thawing to release intracellular components. The supernatants were collected carefully after centrifuging for 20 min at 2,000 rpm. Intracellular acetylcholine levels were measured by using a commercially available ELISA kit. (Acetylcholine ELISA Kit; Biovision, Milpitas, CA, USA). Absorbances were measured at O.D. 450 nm.
Neurite elongation
For determination of neurite elongation, HT22 cells instead of N2a cells were cultured in the six-well culture plate at a density of 1,000 cells per cm2 on the cover slips [29]. HT22 cells were incubated with Aβ1-42 (3 μM) alone or with donepezil, CA and CAD for 5 days. For the analysis, cells were fixed in the paraformaldehyde (4%) and then incubated with SMI-312 antibody (Cat. No. SMI312R; Covance, Princeton, NJ, USA) for 1 h. Secondary antibody conjugated to Alexa Fluor 488 (Invitrogen, Carlsbad, CA, USA) was applied for 1 h. Fluorescent images were magnified at ×400 using an Axiovert 200 fluorescence microscope (Carl Zeiss, Jena, Germany). K252A was from Calbiochem (San Diego, CA, USA) and imatinib was from Toronto Research Chemicals (Toronto, ON, Canada). Methyllycaconitine citrate was purchased from Research Biochemicals International, Natick, MA and LY294002, was from Calbiochem, Darmstadt, Germany.
Cell viability
Cell viability was evaluated by a modified MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma-Aldrich] reduction assay. Cells were pretreated with drugs for 3 h before adding Aβ1-42 (10 μM). Then, 10 μl of MTT (at 5 mg/ml in phosphate-buffered saline was included to each well (500 μg/ml) for 3 h. Optical densities were measured at a wavelength 570 nm (reference wavelength 630 nm) using enzyme-linked immunosorbent assay (Bio-Tek Instruments, Burlington, VT, USA).
Statistical analyses
Results were expressed as means ± standard error of the mean. The significances of differences between control and drug treatment groups were analyzed by ANOVA followed by Tukey’s multiple comparison test. The Student’s t-test was used to determine the significances of differences between untreated cells and cells treated with inhibitors. p < 0.05 was judged statistically significant.
RESULTS
Time-dependent increases in amyloid-β peptide with P-tau, Ac-tau, P300 and GSK-3β P-Tyr-216 levels in the activated N2a Swe cells
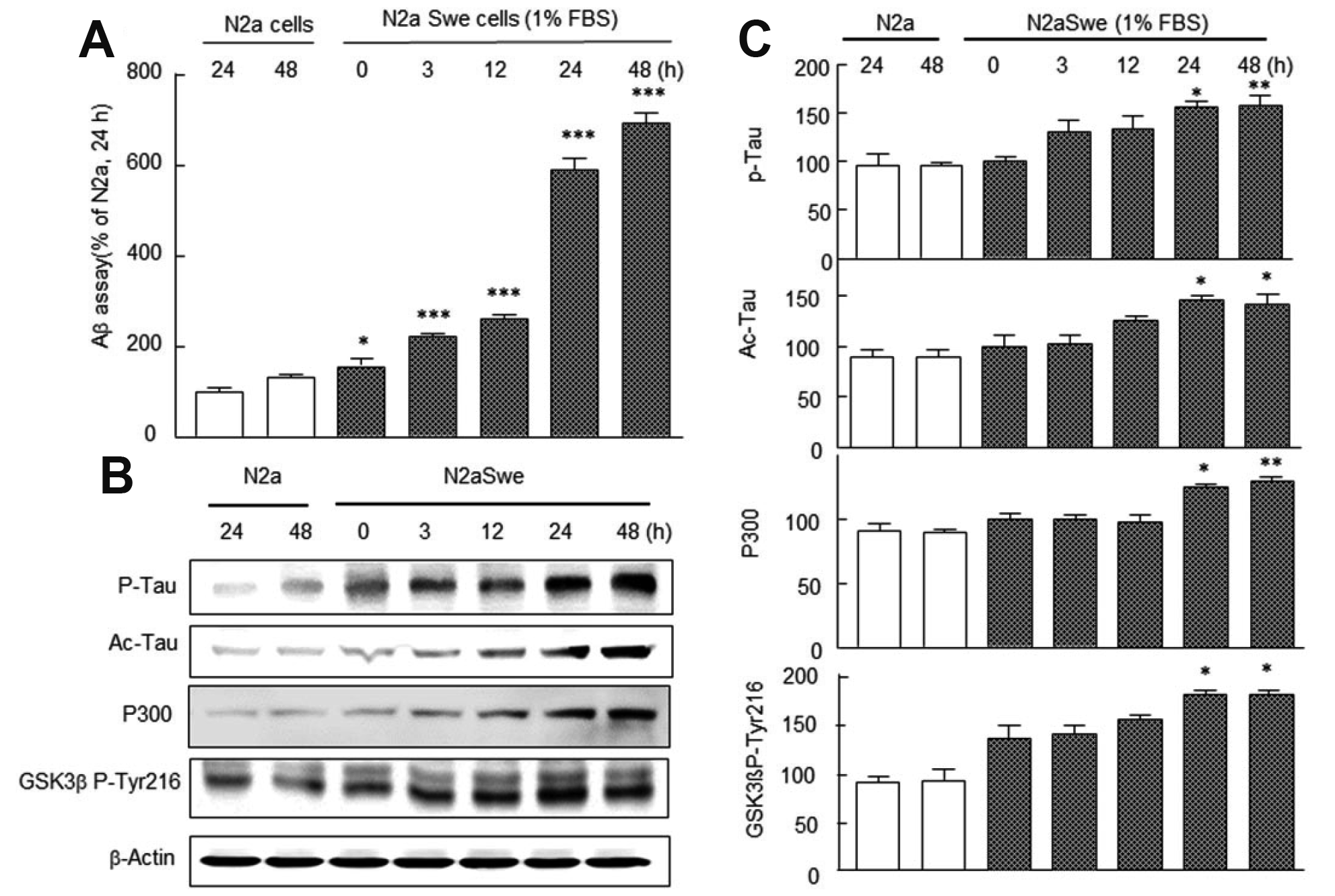
We assayed time-dependent increases in Aβ, phosphorylated tau (P-tau), acetyl tau (Ac-tau), P300, and GSK-3β P-Tyr-216 expression levels by ELISA in the activated N2a Swe cells. To induce Aβ overproduction, culture medium was switched from medium containing 10% FBS to medium with 1% FBS, and neuronal cells were cultured for 3, 12, 24, and 48 h. As shown in Fig. 1A, N2a cells showed little Aβ density before or at 24–48 h after FBS depletion. Aβ levels (assayed by ELISA) were also low in N2a cells after culture for 24–48 h in FBS depleted medium. However, in the activated N2a Swe cells Aβ concentrations increased time-dependently (3, 12, 24, and 48 h), and the higher concentration was observed from 24 h (591 ± 24.9%) (Fig. 1A). In addition, Western blotting for P-tau (F6,20 = 8.483, p = 0.0005), Ac-tau (F6,20 = 9.19, p = 0.0003), P300 (F6,20 = 10.99, p = 0.0001), and GSK-3β Tyr216 (F6,20 =12.49, p = 0.0001) also showed time-dependent increases that paralleled those of Aβ levels (Fig. 1B).
Inhibition of increased Aβ, P-tau, Ac-tau, P300, and GSK-3β P-Tyr-216 levels in activated N2a Swe cells by CAD
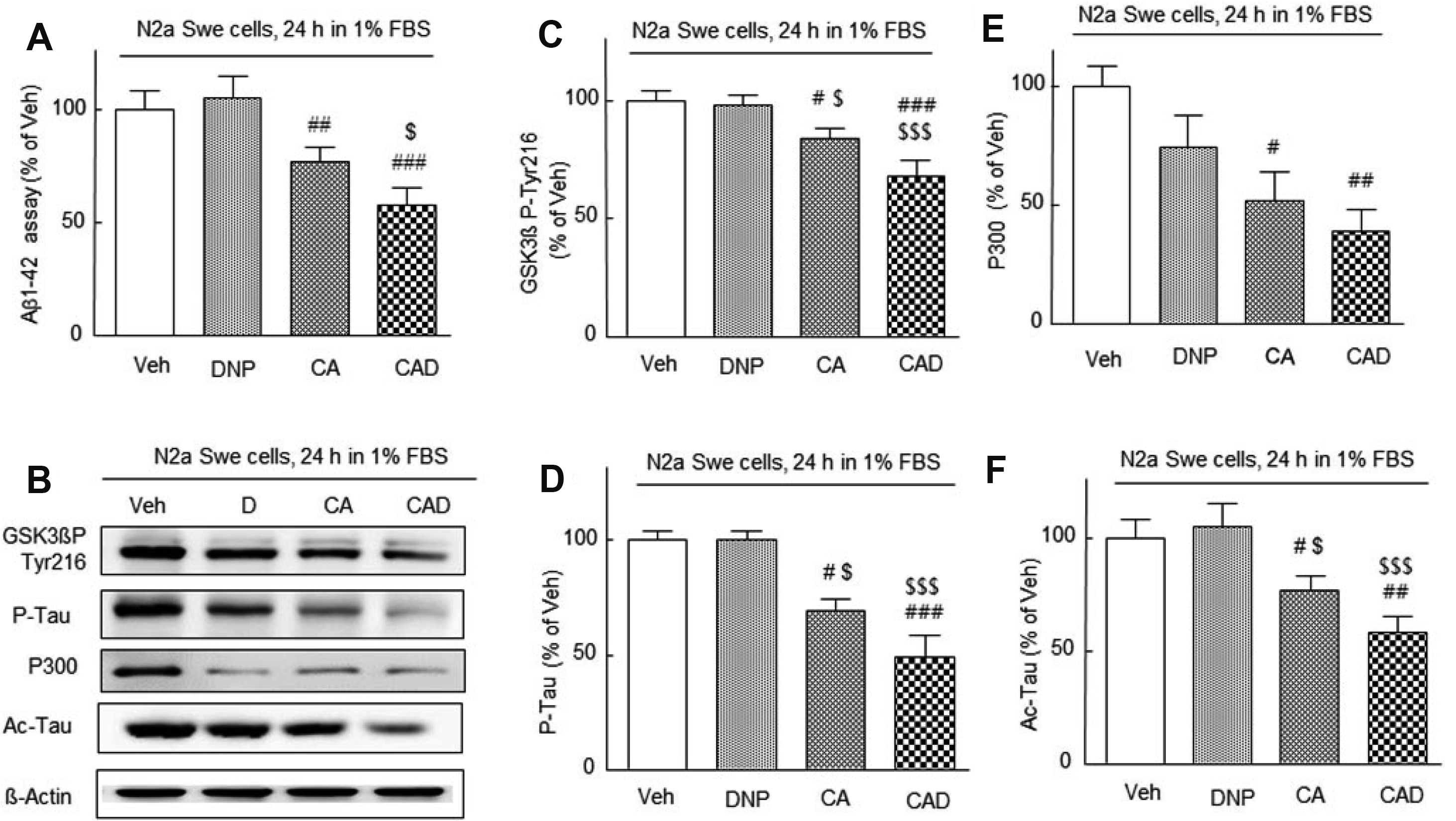
It was aimed to evaluate whether the three-drug combination (CAD) therapy could synergistically suppress endogenously accumulated Aβ level in activated N2a Swe cells. As observed in the previous reports [29,35], minimum concentrations of 3 μM of cilostazol and 3 μM of aripiprazole were applied in combination in this study. N2a Swe cells were pretreated with drugs 3 h prior to exposure to 1% FBS medium and then cultured for further 24 h. Aβ accumulation was not suppressed by 3 μM DNP alone but was significantly suppressed by CA (cilostazol 3 μM + aripiprazole 3 μM, p < 0.01) and further suppressed by CAD (CA + DNP) (p < 0.001). CAD more strongly suppressed Aβ accumulation (to 55.0 ± 6.5%, p < 0.05) than CA (to 65.0 ± 3.9%), indicating that Aβ accumulation was more efficiently decreased by treatment with CAD (Fig. 2A).
It has been reported that phosphorylation of GSK-3β at Tyr 216 increases GSK-3β activity in neuronal cells and in the ischemic cortical neurons [36]. Furthermore, active GSK-3β increased hyperphosphorylation of tau [37]. Based on these reports, we examined whether CAD therapy could synergistically suppress GSK-3β P-Tyr-216, P-tau, P300, and Ac-tau expression in parallel with reduced Aβ level (Fig. 2B).
FBS-depleted medium-induced increased GSK-3β Tyr216 was not inhibited by DNP, but both CA and CAD treatment significantly suppressed this increase to 73 ± 12.3% (p < 0.05) by CA and to 54 ± 10.0% (p < 0.001) by CAD, respectively (F3,15 = 16.9, p = 0.0001). Similarly, P-tau expression were also not affected by DNP but were significantly suppressed by CA (69 ± 5.6%, p < 0.05 vs. vehicle) and CAD (49.0 ± 9.5%, p < 0.001) (F3,15 = 15.9, p = 0.0002) (Fig. 2C, D).
Aβ aggregates have been reported to activate p300 (protein of 300 kDa), and tau is acetylated by histone acetyltransferase in an intracellular Aβ accumulation-dependent manner [38]. Recently, it was reported that acetylated tau inhibits its degradation and enhances tau aggregation [39]. P300 expressional increases in N2a Swe cells after 24 h in depleted medium were not inhibited by DNP, but were significantly inhibited by CA (52 ± 12.0%, p < 0.05 vs. vehicle) and CAD (39 ± 9.6%, p < 0.01) (F3,15 = 5.697, p = 0.0116) (Fig. 2E, F), and changes in Ac-tau levels displayed a similar pattern (F3,15 = 14.66, p = 0.0003). These results indicate that CAD treatment synergistically inhibited the expressions of several neurodegenerative pathological factors which were induced by endogenous Aβ accumulation.
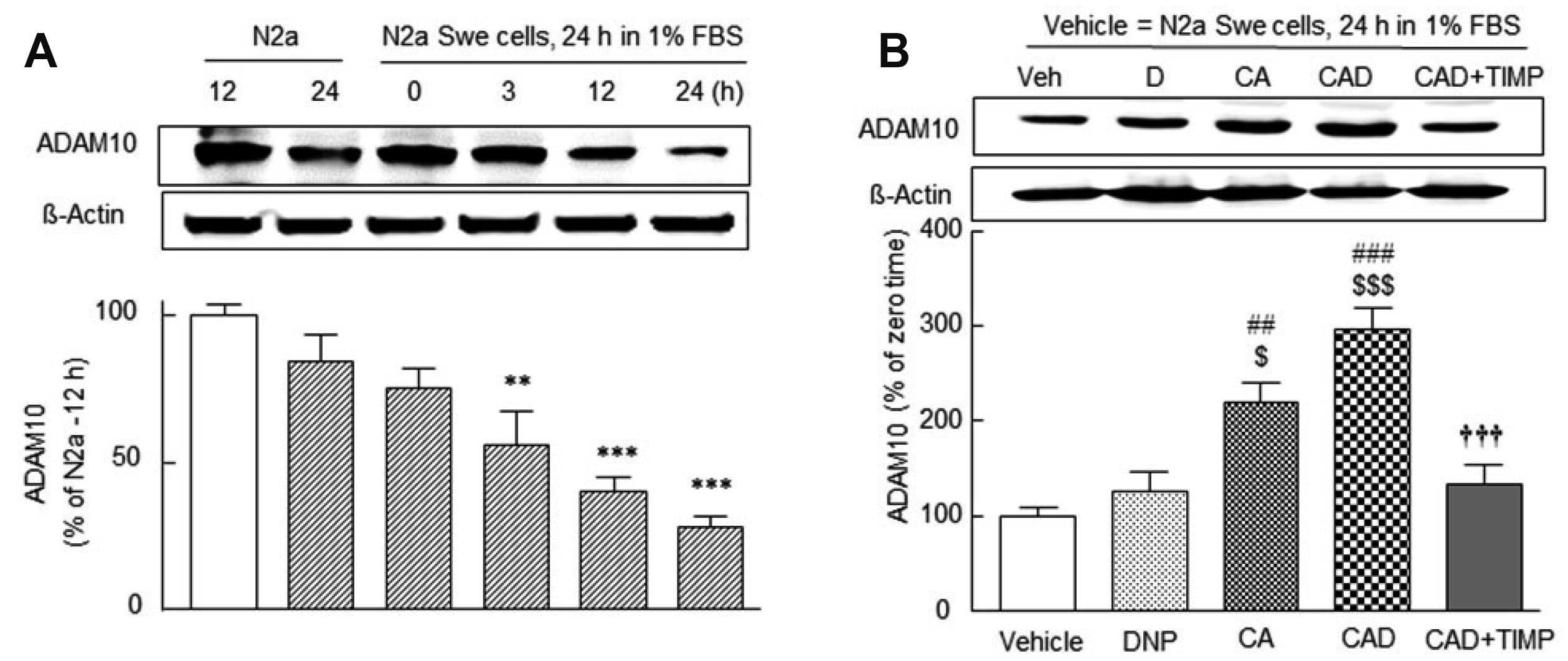
Time-dependent decrease in ADAM10 expression in N2a Swe cells in FBS-depleted medium and synergistic upregulation of ADAM10 by CAD
Postina et al. [3] reported that neuronal overexpression of ADAM10 augments the α-secretase cleavage of APP and its activation inhibits Aβ peptide production via APP cleavage. Lee et al. [19] have shown that cilostazol attenuates Aβ production by increasing ADAM10/α-secretase activity via SIRT1-coupled RARβ activation in N2a Swe cells. As shown in Fig. 3A, ADAM10 expression significantly decreased in N2a Swe cells in FBS-depleted medium in a time (0, 3, 12, and 24 h)-dependent manner (F5,23 = 14.60, p < 0.0001), while it remained unchanged in N2a cells. Moreover, ADAM10 expression after 24 h in depleted FBS medium (28.3 ± 4.3% of the N2a cells, control = 100%) was increased by CA pretreatment to 220 ± 20.8% (p < 0.01 vs. vehicle) and by CAD to 297 ± 21.8% (p < 0.001 vs. activated N2a Swe) (F4,19 = 17.7, p < 0.0001). The protective effect of CAD was suppressed by 10 μM TIMP (an ADAM10 inhibitor, Amour et al. [40]) to 133 ± 21.1% (p < 0.001 vs. activated N2a Swe controls) (Fig. 3B).
Decrease in α-secretase activity in the activated N2a Swe cells and its upregulation by CAD therapy
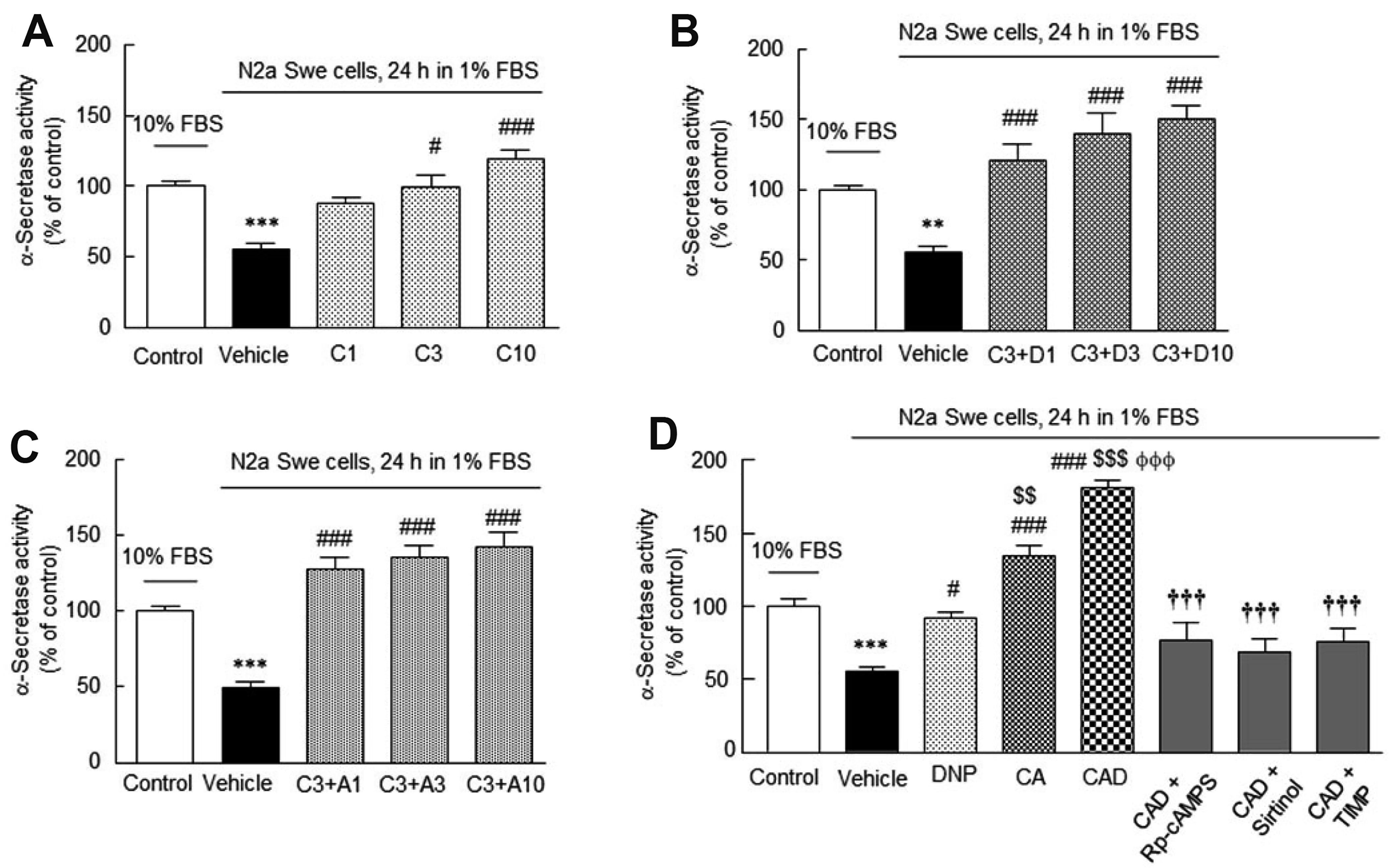
We investigated how cilostazol, cilostazol + DNP, cilostazol + aripiprazole, and CAD affect α-secretase activity in the activated N2a Swe cells. α-Secretase activity was significantly reduced to 55 ± 4.6% after 24 h in depleted medium (p < 0.001). This reduction was significantly prevented by pretreatment with 3 μM (C3) and 10 μM (C10) of cilostazol to 99.0 ± 8.6% (p < 0.05) and 119.2 ± 6.0% (p < 0.001), respectively. α-Secretase activity was also significantly elevated by cotreatments with cilostazol (3 μM) and DNP (1, 3, 10 μM), or by cotreatments with cilostazol (3 μM) and aripiprazole (1, 3, 10 μM) (Fig. 4A–C).
We also compared the abilities of DNP alone, CA and CAD to upregulate α-secretase activity in the activated N2a Swe cells. In this experiment, reduced α-secretase activity (56. 1 ± 2.7%, p < 0.001) was synergistically elevated to 181.3 ± 5.6% by CAD, which was much higher than those achieved by DNP alone or cilostazol + aripiprazole combination (F7,43 = 40.9, p < 0.0001). This upregulation by CAD was significantly inhibited by pretreatment with Rp-cAMPS (10 μM, a cAMP-dependent protein kinase inhibitor) to 77.0 ± 11.5% (p < 0.001), by pretreatment with sirtinol (20 μM, SIRT1 inhibitor) [41] to 69.5 ± 9.2% (p < 0.001), and by TIMP (10 μM, an ADAM10 inhibitor) [40] to 76.3 ± 8.5% (p < 0.001) (Fig. 4D).
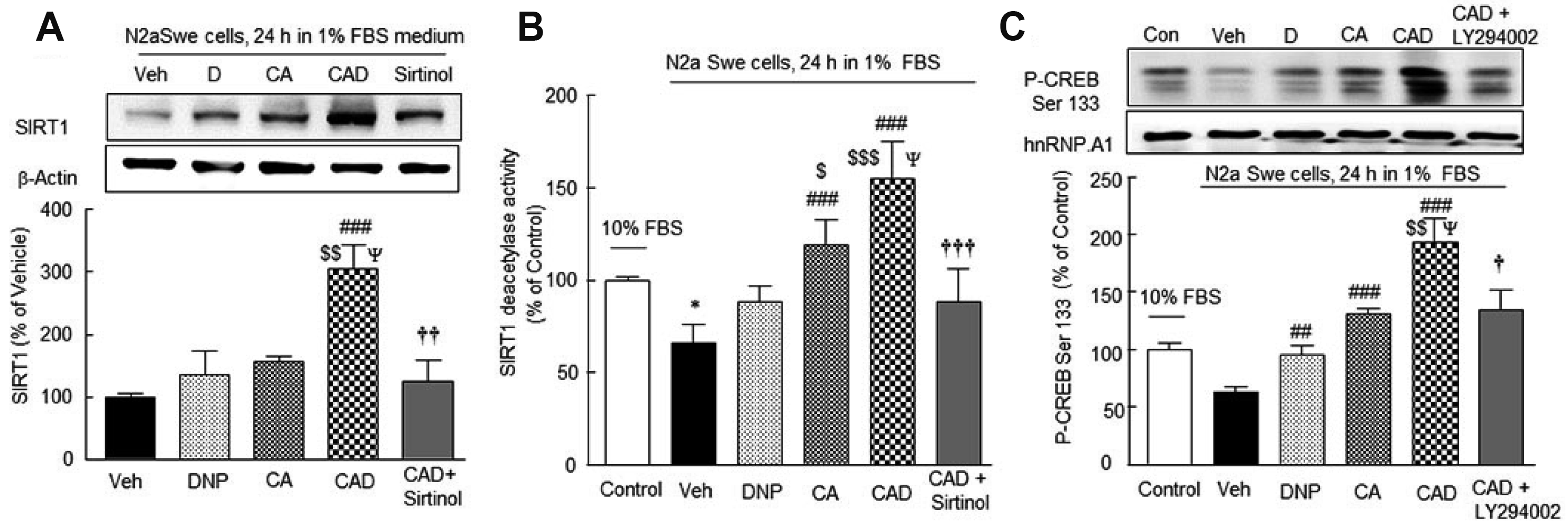
CAD treatment synergistically increased SIRT1 protein expression and its activity
Lee et al. [35] reported that in the activated N2a Swe cells, SIRT1 protein expression was time (3, 12, 24, 48 h)-dependently decreased. In the present study, N2a Swe cells were pretreated with 3 μM DNP, CA (3 μM cilostazol + 3 μM aripiprazole), or CAD (3 μM cilostazol + 3 μM aripiprazole + 3 μM DNP) for 3 h, and then cultured in 1% FBS medium for 24 h. Results were expressed as percentages of activated N2a Swe controls (vehicle = 100%). SIRT1 expression was significantly upregulated by CAD to 304.6 ± 38.2% (p < 0.001 vs. vehicle; p < 0.01 vs. DNP; p < 0.05 vs. CA) in a synergistic manner (F4,19 = 9.221, p = 0.0006), whereas it was marginally changed by 3 μM DNP and CA (3 μM cilostazol + 3 μM aripiprazole). Furthermore, this effect of CAD was significantly inhibited by 20 μM sirtinol (p < 0.01, a SIRT1 inhibitor) (Fig. 5A).
SIRT1 deacetylase activities were significantly reduced to 66 ± 4.5% (p < 0.05) and this reduction was significantly prevented by CA to 119 ± 6.1% (p < 0.001 vs. vehicle) and more increased by CAD to 155 ± 9.0% (p < 0.001 vs. vehicle; p < 0.001 vs. DNP; p < 0.05 vs. CA) (F5,29 = 19.77, p = 0.0039), and these effects were significantly suppressed by 20 μM sirtinol (p < 0.001), indicating that SIRT1 activity was decreased by intracellular Aβ, and three-drug combination therapy (CAD) upregulated the reduced SIRT1 protein expression and deacetylase activity in a synergistic manner (Fig. 5B).
Fusco et al. [42] have reported that CREB directly upregulates SIRT1 mRNA and protein expression in neurons. In the present study, CREB and P-CREB Ser133 expression was reduced by endogenously produced Aβ in FBS-depleted medium in N2a Swe cells and that CAD synergistically reversed this decrease (F5,17 = 13.09, p = 0.0002) and this increased P-CREB Ser133 level by CAD was significantly depressed by LY294002 (PI3-kinase inhibitor) (F5,17 = 13.09, p = 0.0002) (Fig. 5C).
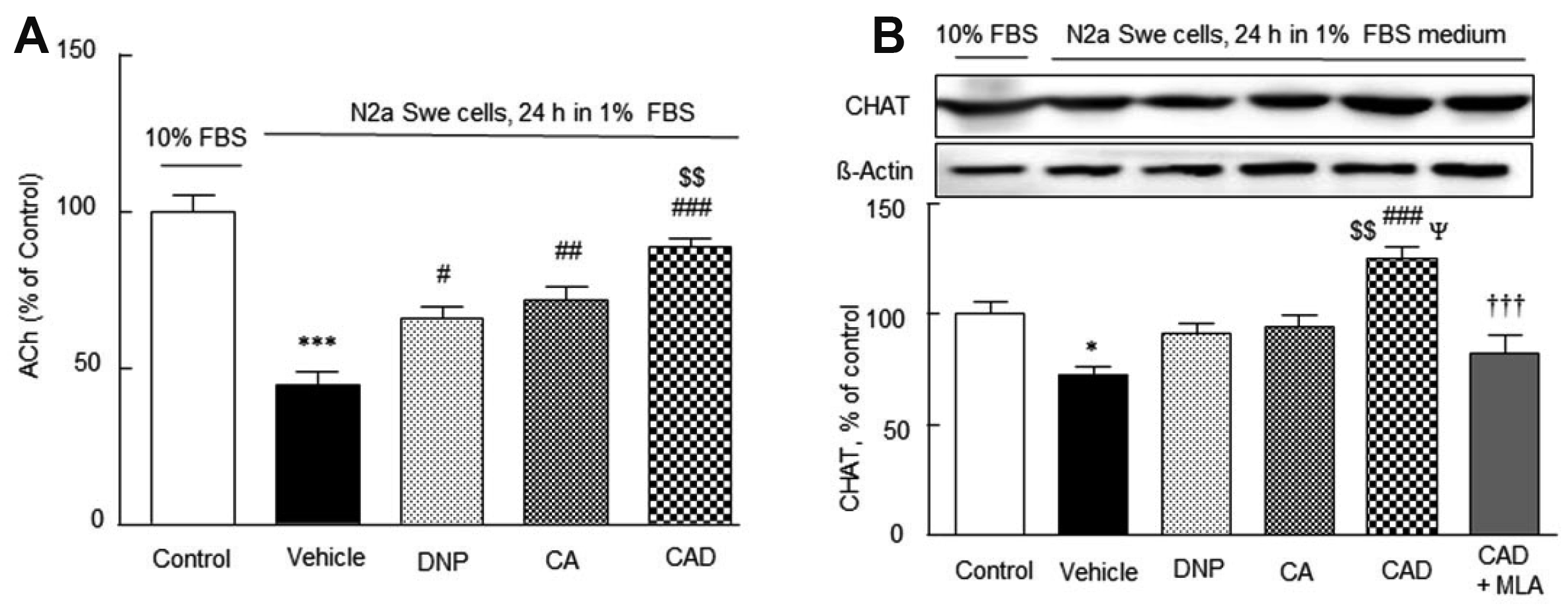
Reductions in acetylcholine release and CHAT expression in the activated N2a Swe cells and overcoming of this cholinergic disruption by CAD
Solubilized amyloid β-peptide inhibits several steps of ACh synthesis/release and inhibits various cholinergic functions at nanomolar concentrations [43] and CHAT expression (a presynaptic marker) [44]. The ACh concentration released to cultured medium containing 10% FBS in the N2a Swe cells was 0.15 ± 0.03 pg/ml (100%) (Fig. 6A), and this was reduced to 45.2 ± 4.0% (p < 0.001) in the activated N2a Swe cells. This decreased concentration was recovered by treatment with DNP (to 66.0 ± 4.7%, p < 0.05), CA (72.3 ± 4.2%, p < 0.01) and by CAD (89.5 ± 2.8%, p < 0.001 vs. vehicle; p < 0.01 vs. DNP) (F4,24 = 25.14, p < 0.0001).
Similarly, CHAT expression was also significantly lowered to 72.2 ± 3.8% (p < 0.05) in the activated N2a Swe cell group, and this reduction was moderately reversed by DNP or CA pretreatment, but significantly recovered over the control level by CAD (to 125.4 ± 5.0%; p < 0.001 vs. vehicle; p < 0.01 vs. DNP; p < 0.05 vs. CA). Furthermore, This CAD-stimulated CHAT expression was significantly blocked by 10 nM methyllycaconitine (MLA) [45], (p < 0.001) (F5,23 = 10.45, p < 0.0001) (Fig. 6B). These results indicate CAD upregulated CHAT expression by activating α7-nicotinic cholinergic receptors in a synergistic manner.
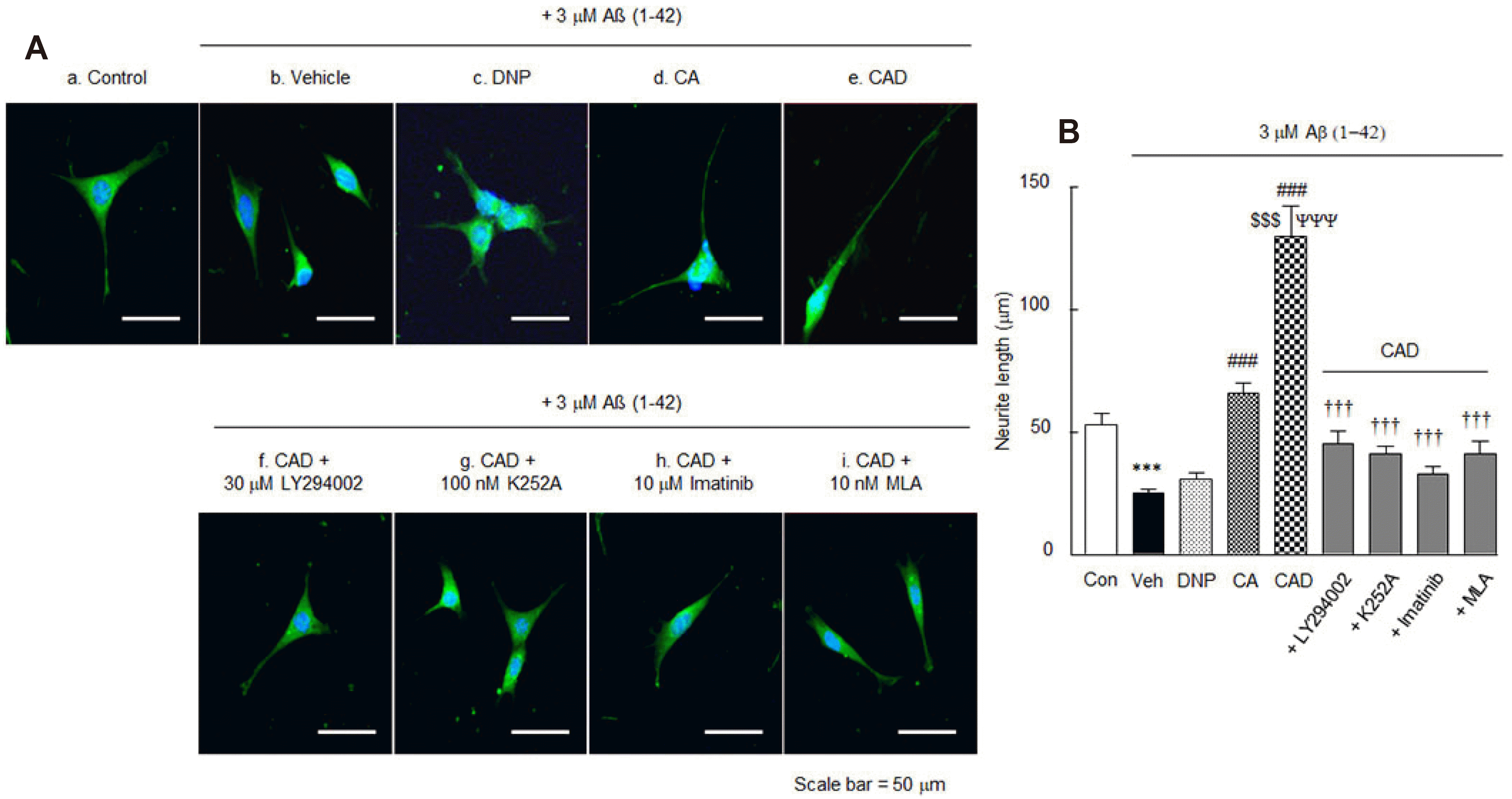
Neurite elongation
Votin et al. [46] have reported that neuronal morphogenesis involves the formation and differentiation of neurites into axons and dendrites. In this study, we used HT22 cells, a murine cell line of hippocampal origin, expressing the BDNF receptor TrkB [47]. The neurite elongation of HT22 cells was analyzed under treatment with DNP alone, CA or CAD for 5 days following incubation with 3 μM of Aβ1-42.
Neurites (control, 53.3 ± 4.5 μm; n = 14) showed significantly short length in Aβ1-42 controls (25.3 ± 1.6 μm, n = 17, p < 0.01), but neurite lengths were significantly increased in CA-pretreated cells (65.9 ± 4.2 μm, n = 23, p < 0.001) and synergistically increased in CAD-pretreated cells (130.0 ± 12.6 μm, n = 7, p < 0.001), but little in DNP-pretreated cells (Fig. 7A). These results strongly indicate that CAD significantly enhanced neurite elongation in HT22 cells.
We also evaluated whether increased neurite elongations induced by CAD were blocked by inhibitors. Increased neurite lengths (130.0 ± 12.6 μm) by CAD were significantly blocked by LY294002 (30 μM) (a PI3K inhibitor) [47] to 45.7 ± 4.8 μm (p < 0.001, n = 22), by K252A (100 nM; inhibitor of TrkB) [48] to 41.0 ± 3.6 μm (p < 0.001, n = 24), by imatinib (10 μM) (10 μM, a β-catenin inhibitor) [49] to 33.3 ± 2.7 μm (p < 0.001, n = 18), and by MLA (10 nM) (methyllycaconitine, a selective inhibitor of α7-nicotinic cholinergic receptor) [50] to 41.1 ± 5.4 μm (p < 0.001, n = 23) (F8,160 = 25.0, p < 0.0001) (Fig. 7B). These results suggest that activations of PI3K, BDNF, β-catenin, and α7-nicotinic cholinergic receptors are involved in the increase in neurite elongation induced by CAD in HT22 cells even in the presence of 3 μM of Aβ1-42.
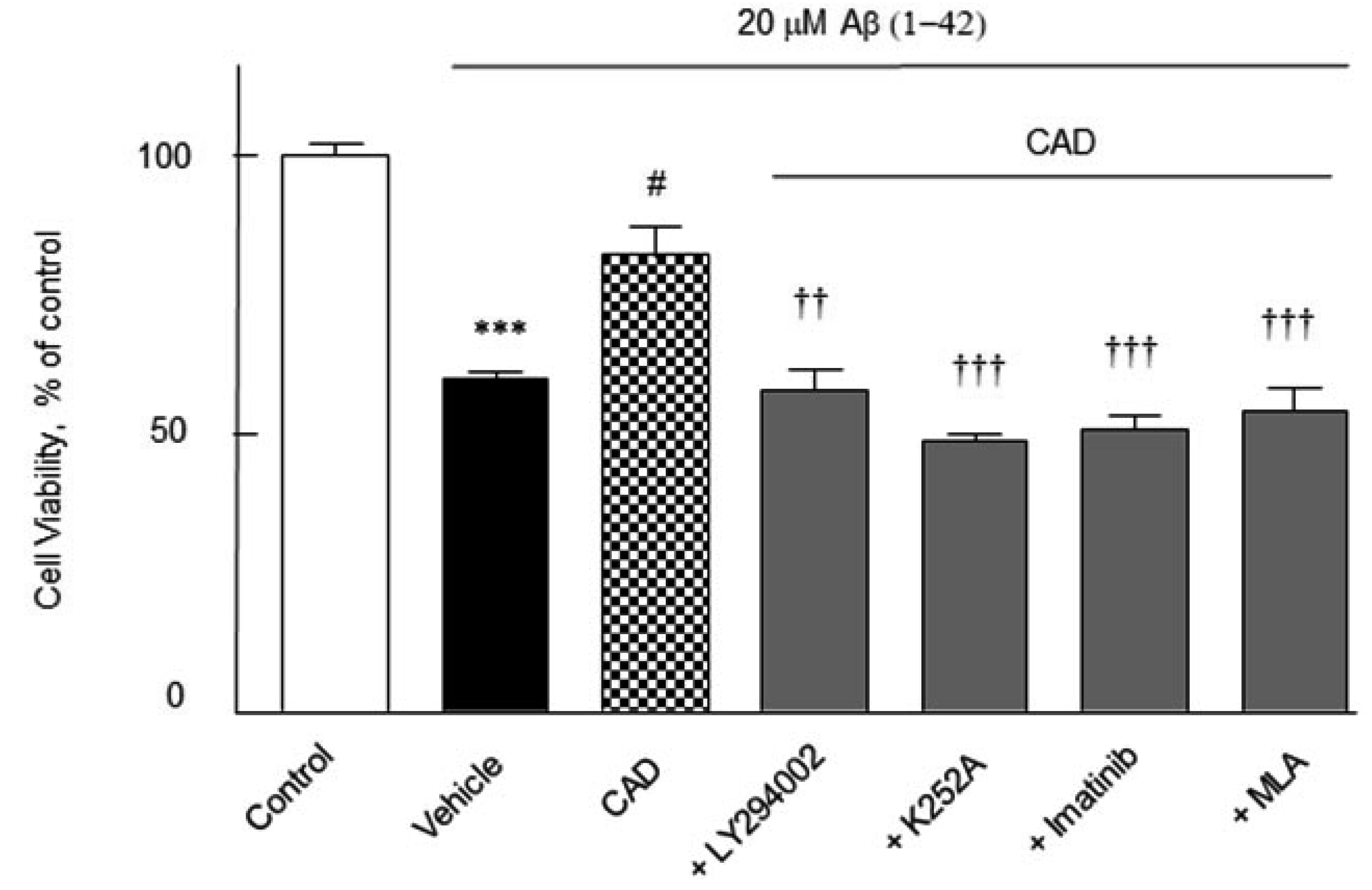
Cell viability
The cell viability of the activated N2a Swe cells was quantified by using a modified MTT reduction assay. N2a cells were pretreated with CAD (3 μM cilostazol + 3 μM aripiprazole + 3 μM donepezil) for 2 h before addition of Aβ1-42 (20 μM) and then cultured forward for 24 h, after which the cell viability was assayed. Upon exposure of N2a cells to Aβ1-42 (20 μM) for 24 h showed a significant decreased cell viability by 60 ± 1.2% (p < 0.001). The decreased viability induced by Aβ1-42 was significantly recovered by CAD to 80.5 ± 5.0% (p < 0.05) at 24 h, which was blocked by 30 μM of LY294002 to 58 ± 3.8% (p < 0.01), 100 nM of K252A to 49.0 ± 1.2% (p < 0.001), imatinib to 51 ± 2.4% (p < 0.001) and 10 nM of MLA to 54 ± 4.4% (p < 0.001) (F6,35 = 7, p < 0.0001) (Fig. 8). These results indicate that activations of PI3K, BDNF, β-catenin and α7-nicotinic cholinergic receptors are importantly involved in the increase in cell viability by CAD even in the presence of 3 μM of Aβ1-42.
DISCUSSION
The present study links upregulation of SIRT1 expression/activity with P-CREB Ser 133 induced by combination therapy with 3 μM cilostazol + 3 μM aripiprazole and 3 μM of donepezil add-on (CAD) to blockade of β-amyloid production (amyloid hypothesis), to inhibition of Ac-tau and P-tau (tau hypothesis) via inhibition of P300 and GSK-3β, and to upregulation of α-secretase/ADAM10 in the activated N2a Swe cells overproducing Aβ. In addition, CAD synergistically upregulated ACh release and CHAT expression in the activated N2a Swe cells. Consequently, CAD treatment synergistically increased neurite elongation and improved cell viability in the presence of Aβ1-42 via activations of PI3K, BDNF, β-catenin, and α7-nicotinic cholinergic receptors in neuronal cells.
A cholinergic hypothesis has been proposed based on the premise that cholinergic agents are a logical choice for preventative AD therapy [13,44]. Nevertheless, single treatment with cholinesterase inhibitors cannot overcome the multifactorial pathologies of AD. Accordingly, we proposed that lower doses of cilostazol plus aripiprazole therapy with add-on donepezil may ensure a superior therapeutic window for the long-term treatment of AD with fewer side effects. In the current study, we identified that CAD exerts synergistic inhibition on the multi-pathogenic factors: Aβ accumulation, P300, Ac-tau, GSK-3β, P-tau by synergistic upregulation of α-secretase/ADAM10 and SIRT1, neuronal ACh content, and CHAT expression.
Tau is acetylated by histone acetyltransferase in the presence of excessive Aβ accumulation [38]. Aβ aggregate has been reported to activate p300 (a 300 kDa protein), which has acetyltransferase activity, and thus, Aβ increases Ac-tau production and tauopathies [39]. In addition, it was reported that phosphorylation of GSK-3β at Tyr 216 increases GSK-3β activity in neuronal cells and degenerate cortical neurons and this activation causes hyperphosphorylation of tau [36,37]. We observed that Aβ accumulation in the activated N2a Swe cells was not suppressed by 3 μM donepezil alone but by CA (p < 0.01) and CAD (p < 0.001). Furthermore, increases in expressions of P300 and GSK-3β P-Tyr216 were not inhibited by 3 μM donepezil or CA but were significantly inhibited by CAD. Consequently, CAD synergistically inhibited P-tau and Ac-tau levels, indicating CAD treatment synergistically inhibit β amyloid accumulation.
A question arises as to the mechanism by which cilostazol elevates SIRT1 expression and activity. Transcription factor CREB was reported to act as a regulator of neuronal differentiation and plasticity [51]. SIRT1 expression is transcriptionally activated by CREB in HepG2 cells stimulated with forskolin [52]. Fusco et al. [42] proposed that effects of calorie restriction on neuronal plasticity and memory are abolished in mice lacking CREB-1 in forebrain and found CREB deficiency drastically reduced SIRT-1 expression. Overall, these results indicate CREB regulates SIRT-1 mRNA and protein expression in neurons. Lee et al. [35] reported that endogenous Aβ overproduction in the N2a Swe cells down-regulated P-CREB Ser133 time-dependently and expression of SIRT1 mRNA and its activity. In the current study, following pretreatment with cilostazol, increase in P-CREB Ser133 level was accompanied by elevated SIRT1 mRNA expression accordingly. Consistent with this report, SIRT1 expression and its deacetylase activity was significantly upregulated by CAD in a synergistic manner, whereas either 3 μM of donepezil or 3 μM cilostazol + 3 μM aripiprazole (CA) had only marginal changes. Furthermore, pretreatment with sirtinol (an SIRT1 inhibitor) significantly inhibited these protective effects of CAD.
On the other hand, Park et al. [29] showed that aripiprazole exhibited preventive effects against Aβ-induced neurotoxicity. They demonstrated aripiprazole decreased BDNF and P-CK2α expression induced by Aβ1-42 were markedly recovered in the N2a cells. In addition, Aβ1-42-induced decreased levels of P-GSK-3β at Ser9 and nuclear P-β-catenin at Ser675 were also recovered by aripiprazole. These results suggest that aripiprazole and cilostazol share some common pharmacological actions for interplay.
Donepezil (AChE inhibitor) elevates ACh levels by inhibiting ACh hydrolysis in the synaptic clefts, and thus, increases cholinergic transmission [53]. In addition, these neuroprotective effects have been ascribed to activation of nicotinic acetylcholine receptors (nAChRs), PI3K/Akt pathway and upregulation of anti-apoptotic Bcl-2 [54-56]. Consistent with these reports, CAD-stimulated CHAT expression in the present study was significantly blocked by MLA (methyllycaconitine, a selective inhibitor of α7-nicotinic cholinergic receptor) [50].
Zimmermann et al. [57] reported that in differentiated human neuroblastoma cells (SH-SY5Y), sAPPα was released into the medium and ADAM10 level was increased in the membrane compartments after donepezil treatment. Postina et al. [3] reported Aβ peptide production is reduced by activating α-secretase. Qin et al. [15] observed activation of mammalian SIRT1 was positively associated with α-secretase activity in vivo, and Lee et al. [19] revealed cilostazol suppresses accumulations of full length-APP and Aβ production by increasing ADAM10 and α-secretase activities via SIRT1-coupled RARβ in N2a Swe cells. In line with these results, we observed SIRT1 protein expression and SIRT1 deacetylase activity were significantly and synergistically upregulated by CAD in the activated N2a Swe cells, but these were marginally increased by DNP or CA. Furthermore, these increases in SIRT1 levels in activated cells were wholly blocked by sirtinol (a SIRT1 inhibitor). Despite the observed lack of efficacy of DNP and CA, the facts that CAD upregulated SIRT1 expression and activity reflects sharing of pharmacological interplay, because DNP has been reported to upregulate SIRT1 protein expression in hippocampi and cortices in senescence-accelerated mouse prone 8 (SAMP8) [58].
In the present study, HT22 cells, mouse hippocampal neuronal cell line, were used instead of N2a cells, because HT22 cells phenotypically resemble neuronal precursor cells expressing BDNF receptor TrkB, and it lacks functional ionotropic glutamate receptors [47]. Thus, it can exclude neurite outgrowth damage by excitotoxicity caused by glutamate other than Aβ1-42. Neurite length which was markedly decreased in the presence of Aβ1-42 was significantly recovered by treatment with CAD in larger degree, indicating CAD synergistically increased neurite elongation in HT22 cells. Furthermore, CAD significantly protected N2a cells from Aβ1-42 -induced cell death. These observations are supported by our previous findings that cilostazol prevented Aβ-induced HT22 apoptosis by downregulating phosphorylated p53 (Ser 15), Bax, and caspase-3 levels, upregulating Bcl-2 levels, and preventing Aβ-induced neurite shortening and decreased cell viability [35]. Furthermore, these protective effects of CAD on neurite shortening and cell death were significantly blocked by LY294002, K252A (an inhibitor of BDNF receptor), imatinib (β-catenin inhibitor), and MLA (methyllycaconitine; a selective inhibitor of α7-nicotinic cholinergic receptor), suggesting that activations of PI3K, BDNF, β-catenin, and α7-nicotinic cholinergic receptors are importantly implicated in the protective effects of CAD on Aβ-induced neurite shortening.
Taken together, the present study shows multitarget-faceted combinatorial drug therapies are worthy of investigation in the contexts of preventing and treating AD, and our results endorse a shift to multitarget -directed therapeutic approaches. In addition, these results support the notion that low dose cilostazol plus aripiprazole with add-on donepezil offers a wider therapeutic window for the long-term treatment of AD.
 XML Download
XML Download