

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Recent cohort studies on the pathophysiological etiology of osteoarthritis (OA) have demonstrated that aging, obesity, and metabolic diseases are closely correlated risk factors for the development of progressive degeneration of articular cartilage in synovial joints [1].
Cholesterol and oxysterol play important roles in physiological processes including development, homeostasis, aging, and even cell death. In addition, the synovial fluid of patients with inflammatory joint conditions is known to contain higher amounts of cholesterol and its derivatives compared to the synovial fluid of normal patients [2]. Furthermore, recent studies showed that cholesterol metabolism is closely associated with the pathogenesis of OA through the regulation of cholesterol synthesis and efflux-related genes [3]. The 25-hydroxycholesterol (25-HC) is an oxysterol that is synthesized from cholesterol by the cholesterol-25-hydroxylase (CH25H), which is encoded by the interferon-stimulated gene CH25H. Recently, Choi et al. [4], reported that the CH25H-CYP7B1 axis was involved in the pathogenesis of OA [4]. Furthermore, recent studies reported that 25-HC induced apoptosis in various types of cells [5-7].
Hence, the aim of the present study was to investigate whether oxysterol 25-HC induced apoptosis in chondrocyte as a pathophysiological catabolic mediator between metabolic syndrome and OA.
METHODS
Preparation and cultivation of chondrocytes
Chondrocytes were isolated from the articular cartilage of rat (5-day-old Sprague-Dawley) knee joints, in accordance with the protocol approved by the Institutional Animal Care and Use Committee (CIACUC2017-A0055) of Chosun University, Gwangju, Korea. The isolated chondrocytes were maintained in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) (ThermoFisher Scientific, Rockford, IL, USA) containing 10% fetal bovine serum (FBS), antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin), and 50 μg/ml ascorbic acid.
Cell viability assay
Chondrocytes (1 × 106 cells/ml) were cultured in 96-well culture plates, and then treated with either interleukin-1β (IL-1β) or 25-HC for 24 h. Following addition of the dimethyl thiazolyl diphenyl tetrazolium salt (MTT) solution, the chondrocytes were further cultured for 4 h. After incubation, the MTT crystals that formed were suspended completely in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) and the absorbance was read at 570 nm using a spectrometer (Epoch microplate spectrophotometer; BioTek, Winooski, VT, USA).
Cell survival assay
Cell survival assay was performed to assess the survival of chondrocytes treated with either IL-1β or 25-HC using a Live/Dead assay kit (Molecular Probes, Carlsbad, CA, USA), which consists of green calcein AM for labeling live cells and ethidium homodimer-1 for labeling dead cells. Chondrocytes were cultured on 8-well chamber slides (Nunc Lab-Tek II Chamber Slide system; Sigma-Aldrich), and then treated with either IL-1β or 25-HC for 24 h. After cultivation, cell survival assay was performed according to the manufacturer’s instructions. Thereafter, the stained cells were imaged using a fluorescence microscope (Eclipse TE200; Nikon Instruments, Melville, NY, USA) and counted to plot the histogram.
Quantitative polymerase chain reaction (qPCR) and quantitative real-time PCR (qRT-PCR)
Chondrocytes were treated with 25 and 50 ng/ml IL-1β for 24 h. Thereafter, total RNA was isolated from the chondrocytes using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The concentration of the isolated total RNA was measured using a Nanodrop 2000 (ThermoFisher Scientific). To synthesize cDNA, 1 µg RNA was reverse transcribed using a ThermoScript reverse transcription-PCR system (Invitrogen) according to the manufacturer’s instructions. For qRT-PCR, cDNA was amplified using an Eco Real-Time PCR system (illumine Inc., San Diego, CA, USA). Relative gene expression was determined using the ΔΔCT method, as detailed by the manufacturer (illumine Inc.). β-actin was used as the internal control. qPCR was performed using 2× TOPsimple DyeMIX-nTaq (Enzynomics, Seoul, Korea) and specific primers on a TaKaRa PCR Thermal Cycler Dice (TaKaRa Bio Inc., Shiga, Japan). Thereafter, the PCR products were electrophoresed on an agarose gel to determine the expression level of the target genes. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. The primer sequences and the conditions used are summarized in Table 1.
Nuclear staining
Chondrocytes were cultured on an 8-well chamber slide and were treated with 0, 10, and 20 µg/ml 25-HC for 24 h at 37°C. Thereafter, the cells were rinsed three times with phosphate buffered saline (PBS; Sigma-Aldrich) at 4°C. The cells were stained with 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich). Nuclear condensation was observed and imaged using a fluorescence microscope.
Fluorescence-activated cell sorting (FACS) analysis
Chondrocytes were cultured in a 6-well plate (SPL Life Science Co. Ltd., Pocheon, Korea), then cells were treated with 0, 10, and 20 µg/ml 25-HC for further 24 h. The chondrocytes were collected, washed with ice-cold PBS, and resuspended in 1× binding buffer (BD Biosciences, San Diego, CA, USA). Thereafter, annexin V-FITC and propidium iodide (PI) (Cell Signaling Technology, Danvers, MA, USA) were added to the chondrocytes and incubated for 15 min at 37°C. Changes in the apoptotic populations were analyzed using a BD Cell Quest version 3.3 (Becton Dickinson, San José, CA, USA).
Western blotting
Chondrocytes were cultured in a 6-well plate and treated with 0, 10, and 20 µg/ml 25-HC for further 24 h. Thereafter, total proteins were extracted from the chondrocytes using a cell lysis buffer (Cell Signaling Technology), according to the manufacturer’s instructions. The protein concentration was determined using a bicinchoninic acid protein assay (ThermoFisher Scientific). Equal amounts of each protein sample were electrophoresed on 10% sodium dodecyl sulfate polyacrylamide gel and subsequently transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Burlington, MA, USA) at 4°C. Thereafter, the PVDF membrane was blocked using 5% (v/v) bovine serum albumin (BSA; Sigma-Aldrich) prepared in Tris buffered saline with Tween 20 (TBS-T) (Santa Cruz Biotechnology Inc., Dallas, TX, USA) and then incubated with the primary antibodies at 4°C. The following primary antibodies purchased from Santa Cruz Biotechnology Inc. and diluted 1:1,000 in TBS-T containing 5% (v/v) BSA were used: antibodies against CH25H (sc-293256), Fas Ligand (FasL; sc-19988), B-cell lymphoma-2 (Bcl-2; sc-7382) , B-cell lymphoma-extra large (Bcl-xL; sc-8392), and Bax-like BH3 protein (Bid; sc-56025). In addition, the following primary antibodies purchased from Cell Signaling Technology and diluted 1:1,000 in TBS-T containing 5% (v/v) BSA were also used: antibodies against caspase-8 (CST#4790), caspase-3 (CST#9662), Poly(ADP-ribose) polymerase (PARP; CST#9542), Bcl-2-antagonist of cell death (Bad; CST#9292), Bcl-2-associated X protein (Bax; CST#2772), caspase-9 (CST#9508), and β-actin (CST#4970). The immunoreactive bands were visualized using the ECL System (Amersham Biosciences, Piscataway, NJ, USA), exposed on radiographic film or MicorChemi 4.2 (Dong-Il Shimadzu Corp., Seoul, Korea).
Immunocytochemistry
Chondrocytes were cultured on 8-well chamber slides, and then treated with 0, 10, and 20 µg/ml 25-HC for further 24 h. Thereafter, the chondrocytes were permeabilized by incubating in PBS containing 0.1% Triton X-100. Then blocking buffer was added to the slide, and then the slide was placed in a humidified chamber and incubated for 2 h. Subsequently, caspase-3 antibody was incubated with the chondrocyte at 4°C overnight. Thereafter, immunohistochemistry was performed using Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s instructions.
Caspase-3/-7 activity assay
Chondrocytes were cultured on 8-well chamber slides, and then treated with 0, 10, and 20 µg/ml 25-HC for further 24 h. Thereafter, the activity of caspase-3/7 was assessed using the cell-permeable fluorogenic substrate PhiPhiLux-G1D2 (OncoImmunin Inc.; Gaithersburg, MD, USA), according to the manufacturer’s instructions, and imaged using fluorescence microscopy.
Histological analysis using safranin-O/fast green staining and immunohistochemistry using caspase-3
25-HC (20 μg/40 μl) was intra-articularly injected into the dissected rat knee joint. Thereafter, rat knee joints were cultured in DMEM/F12 containing 10% FBS for 14 days. At the end of the culture period, rat knee joints were fixed in 4% paraformaldehyde for 72 h and decalcified in ethylenediaminetetraacetic acid (Sigma-Aldrich). Thereafter, the samples were embedded in paraffin and were serially sliced to 5 μm thickness and placed on slides. Safranin-O/fast green staining was subsequently performed to assess proteoglycan loss in the articular cartilage ground substance. Immunohistochemistry using caspase-3 was performed using Vectastain ABC kit according to the manufacturer’s instructions.
RESULTS
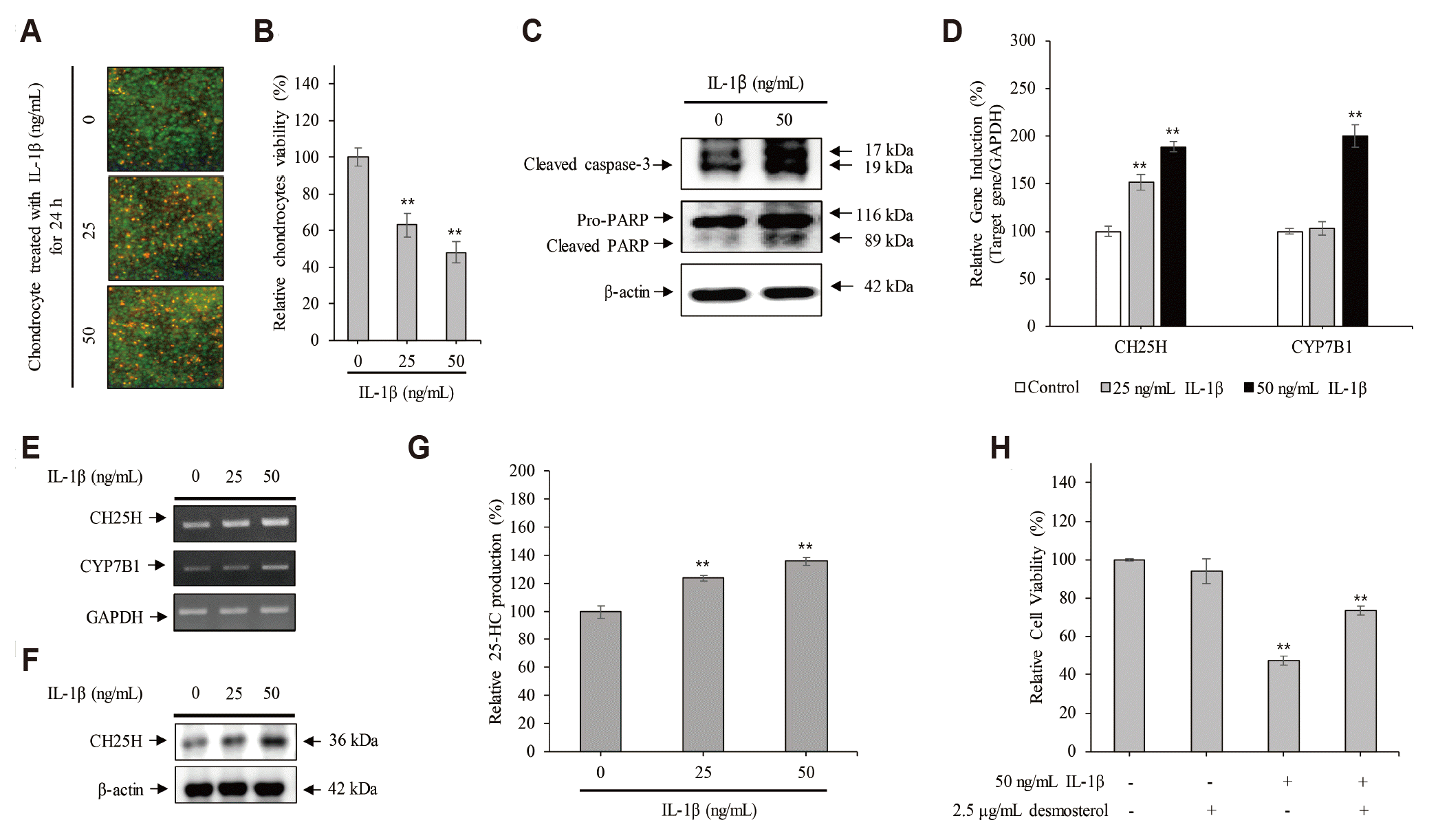
IL-1β-induced apoptotic chondrocyte death is involved partially with cholesterol metabolism
The results of Live/Dead assay showed that the dead cells (that stained red fluorescence) were found to be 63 ± 8% and 48 ± 5% in the chondrocytes treated with 25 and 50 ng/ml IL-1β, respectively, compared with control (98 ± 7%) (Fig. 1A, B). Furthermore, the expression of cleaved caspase-3 and its downstream target PARP increased significantly in the chondrocytes treated with 50 ng/ml IL-1β for 24 h, compared to that in the untreated control cells (Fig. 1C). These data indicate that severe inflammation induces apoptotic chondrocyte death.
Next, to verify whether the IL-1β-induced apoptotic chondrocyte death was associated with cholesterol metabolism, chondrocytes were cultured with 25 and 50 ng/ml IL-1β for 24 h. As shown in Fig. 1D and he level of the CH25H mRNA, coding for an enzyme that converts cholesterol to 25-HC, was increased significantly in chondrocytes treated with IL-1β in a dose-dependent manner. Furthermore, the level of CYP7B1 mRNA, coding for an enzyme that converts 25-HC to 25-hydroxycholesterol 7α-hydroxylase, was increased dose-dependently by IL-1β in chondrocytes. Moreover, the expression of CH25H was also increased in the chondrocytes treated with IL-1β (Fig. 1F). Sequentially, the production of 25-HC was significantly increased to 124 ± 2.08% and 135.9 ± 2.84% in chondrocytes treated with 25 and 50 ng/ml IL-1β for 24 h, respectively, compared to the control cells (100 ± 4.3%) (Fig. 1G). However, to verify whether IL-1β-induced apoptotic chondrocyte death is involved with cholesterol metabolism, chondrocytes were treated with 2.5 μg/ml desmosterol, a potential inhibitor of CH25H [8,9], in the presence or absence of 50 ng/ml IL-1β for 24 h. The results of MTT assay showed that desmosterol was significantly counteracted the IL-1β-induced apoptotic chondrocyte death (Fig. 1H).
Therefore, these data indicate that IL-1β-induced apoptotic chondrocyte death is involved partially with cholesterol metabolism.
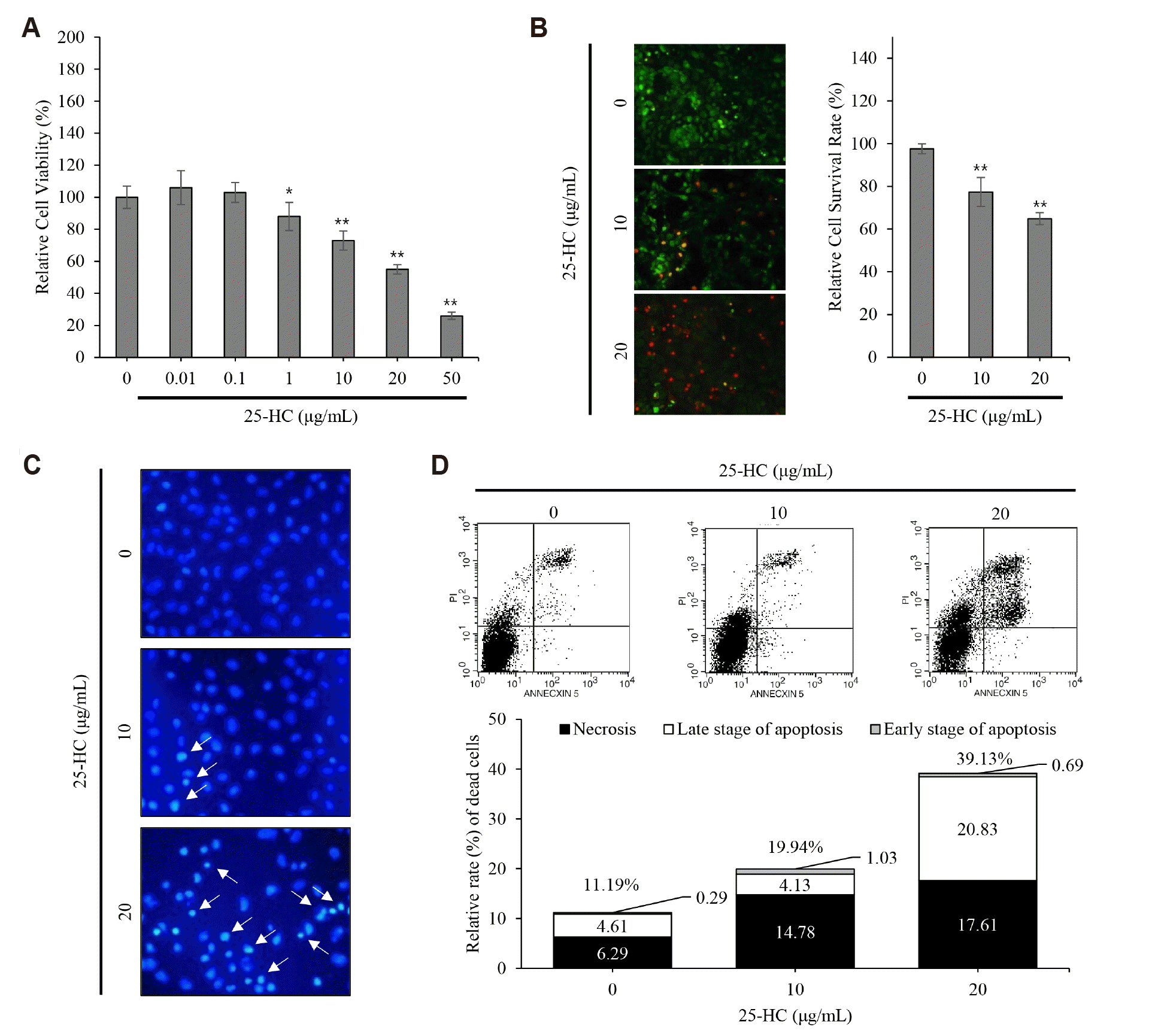
25-HC induces the apoptotic chondrocytes death
The results of MTT assay showed that relative cell viabilities were decreased to 73 ± 6% and 55 ± 3% in the chondrocytes treated with 10 and 20 μg/ml 25-HC for 24 h, respectively, compared to the control (100 ± 7%) (Fig. 2A). Furthermore, the results of Live/Dead assay showed that the number of dead chondrocytes stained with red fluorescence was increased in an IL-1β dose-dependent manner and relative cell survival rates were found to be 77.38 ± 6% and 64.85 ± 3% in the chondrocytes treated with 10 and 20 μg/ml 25-HC for 24 h, respectively, compared to the control (97.6 ± 4%) (Fig. 2B). Therefore, these data indicate that 25-HC consistently acts as a toxic oxysterol in chondrocytes.
Next, to investigate whether the 25-HC-induced chondrocyte death was associated with apoptosis, nucleus condensation, a characteristic feature of apoptosis, was assessed in the chondrocytes treated with 10 and 20 μg/ml 25-HC for 24 h using DAPI staining. The number of chondrocytes with condensed nucleus increased following 25-HC treatment in a dose-dependent manner (Fig. 2C). FACS analysis showed that the relative rate of cell death was 19.94% and 30.13% in the chondrocytes treated with 10 and 20 μg/ml 25-HC, respectively, compared to the control cells (11.19%) (Fig. 2D). Furthermore, among the dead chondrocytes, the relative rate of apoptotic cell death including early and late stage of apoptosis was 4.9%, 5.16%, and 21.52% in the chondrocytes treated with 0, 10, and 20 μg/ml 25-HC, respectively. Taken together, these data indicate that the 25-HC-induced chondrocyte death is mediated by apoptosis.
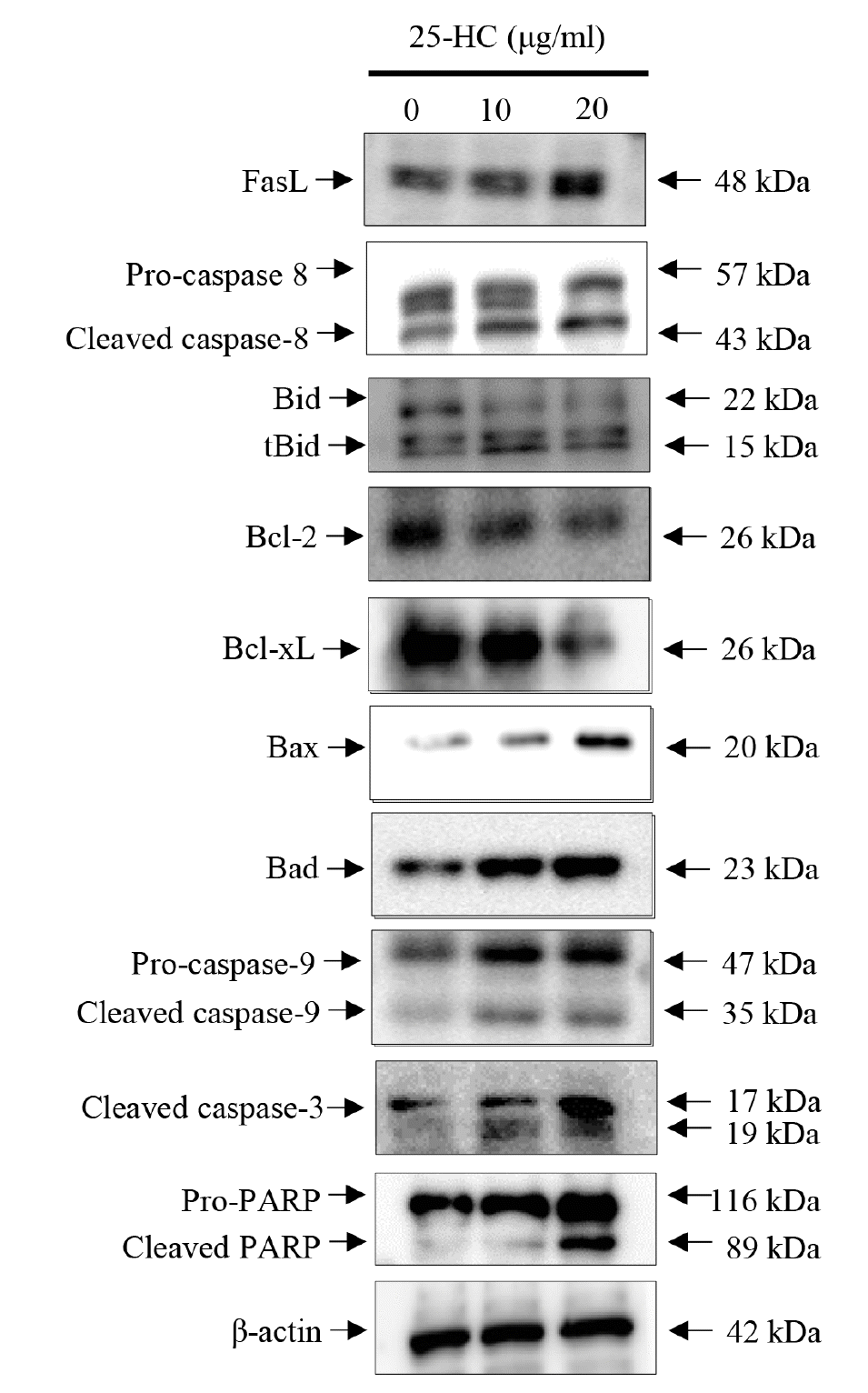
25-HC induces death receptor-mediated extrinsic and mitochondria-dependent apoptosis in chondrocytes
As shown in Fig. 3, the expression of FasL, a death ligand associated with death receptor-mediated extrinsic apoptosis, increased dose-dependently in the chondrocytes treated with 25-HC for 24 h. Furthermore, the expression of pro- and cleaved caspase-8, a downstream target of FasL, increased dose-dependently by 25-HC. Sequentially, 25-HC induced chondrocyte death through the expression of cleaved caspase-3, pro- and cleaved PARP. Hence, these data indicate that the 25-HC-induced chondrocyte death is mediated by death receptor-mediated extrinsic apoptosis.
Cleaved caspase-8 induces the cleavage of Bid, a pro-apoptotic factor associated with mitochondria-dependent apoptosis, to tBid. The expression of tBid (truncated/cleaved Bid) mediated by cleaved caspase-8 increased dose-dependently in the chondrocytes treated with 25-HC. Furthermore, the expression of Bcl-2 and Bcl-xL, known as an anti-apoptotic factors in mitochondria-dependent apoptosis, decreased while the expression of pro-apoptotic factors such as Bax, Bad, pro- and cleaved caspase-9 increased to initiate the cleavage of pro-caspase-3 in the chondrocytes treated with 25-HC. Sequentially, 25-HC induced chondrocyte death through the expression of cleaved caspase-3 and cleaved PARP. Therefore, these data indicate that the 25-HC-induced chondrocyte death is mediated by the expression of caspase-3 through death receptor-mediated extrinsic and mitochondria-dependent intrinsic apoptotic pathways.
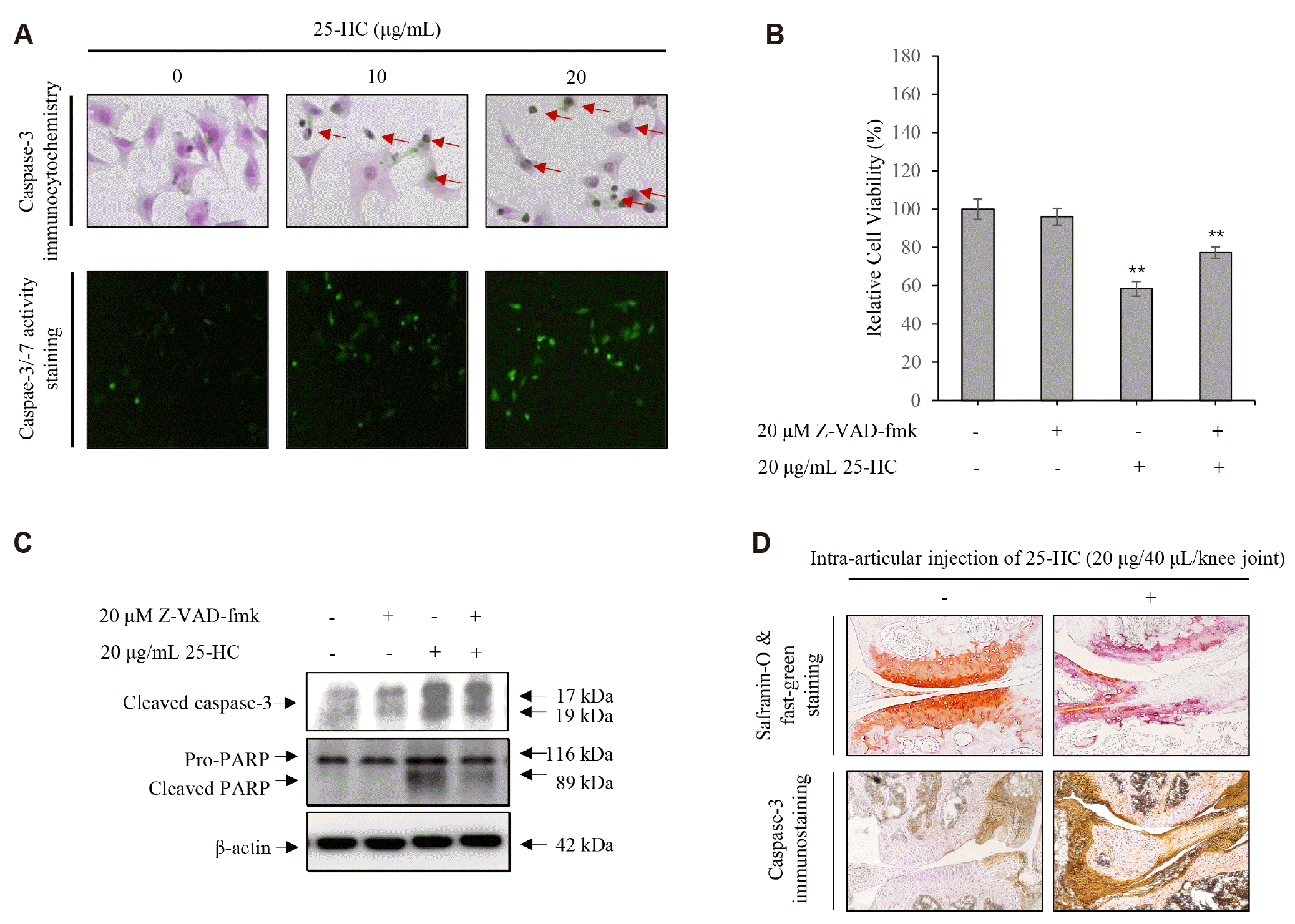
25-HC induces not only caspase-dependent apoptosis, but also induces proteoglycan loss in articular cartilage
The results of immunohistochemistry and caspase-3/-7 activity assay to investigate the expression and activation of caspase-3 in the chondrocytes treated with 25-HC showed that the chondrocytes with shrinking morphology were not only increased by 25-HC, but the expression of caspase-3 also increased in these chondrocytes (Fig. 4A). Furthermore, the activity of caspase-3 increased in the chondrocytes treated with 25-HC (Fig. 4B). To further verify whether the 25-HC-induced apoptosis was caspase dependent, chondrocytes were stimulated with 20 μg/ml 25-HC in the presence or absence of 20 μM Z-VAD-fmK (Santa Cruz Biotechnology Inc.), a pan-caspase inhibitor, for 24 h. The results of MTT assay showed that the relative viability of chondrocytes was 96 ± 4% in presence of Z-VAD-fmk compared to the control (100 ± 5%). However, although 25-HC decreased the relative viability of chondrocytes by 58 ± 4%, it was rescued significantly (77 ± 3%) in the presence of Z-VAD-fmk, as shown in Fig. 4B. Furthermore, the 25-HC-induced caspase-3 expression was significantly counteracted by Z-VAD-fmk treatment in the chondrocytes (Fig. 4C). Moreover, the results of safranin-O/fast green staining showed that intra-articular injection of 25-HC induced severe proteoglycan loss, a typical characteristic of articular cartilage degeneration, compared to the control (Fig. 4D). Furthermore, the immunoreactivity of caspase-3 increased significantly in the articular cartilage of the knee joint following intra-articular injection with 25-HC. Hence, these data suggest that 25-HC not only induces caspase-dependent apoptosis, but also induces proteoglycan loss in the articular cartilage.
DISCUSSION
High levels of cholesterol in the body are one of the metabolic risk factors [10]. Cholesterol, which is generally produced by the cellular cholesterol biosynthetic pathway or dietary intake, is an essential molecule in maintaining various cellular functions including as an essential constituent of cell membranes, a precursor for steroid hormones, and the synthesis of biliary salt. Interestingly, the concentrations of cholesterol and cholesterol crystals in the synovial fluid of patients with OA are higher than that of normal individuals [2]. However, the mechanism responsible for the increase in cholesterol and its physiological role in the synovial fluid has not yet been explored. Recently, Choi et al. reported that cholesterol metabolism mediated by CH25H-CYP7B1-RORα (Retinoic acid receptor-related receptor alpha) regulates OA [4]. They showed that cholesterol levels in chondrocytes not only increased in OA and inflammatory circumstances, but the pathogenesis of OA was also mediated by the upregulation of the cartilage degrading enzymes through the expression of CH25H and production of its metabolite, 25-HC [4]. However, recent studies reported that 25-HC induced caspase-dependent apoptosis in various types of cells such as keratinocytes [5], vascular smooth muscle cells in experimental hypercholesterolemia [11], oligodendrocytes [7], PC12 cells [12], and adult rat Lydig cells [13]. Hence, we hypothesized that 25-HC, an oxysterol produced by CH25H, might be closely associated with the apoptosis of chondrocytes and the pathological link between increase in cholesterol amount in OA and a low-grade chronic inflammation generated synergistically by aging-induced inflammaging, and metabolic inflammation accompanying metabolic syndrome.
Apoptosis of chondrocytes clearly occurs in OA and is the main pathogenic mechanism in OA progression [14]. In present study, we demonstrated that inflammation increased the production of 25-HC through the expression of CH25H in chondrocytes. Furthermore, 25-HC decreased the viability of chondrocytes. Moreover, 25-HC not only induced the death receptor-mediated extrinsic apoptosis that was triggered by the expression of FasL in the chondrocytes, but also induced the mitochondria-dependent intrinsic apoptosis. In addition, both death receptor-mediated extrinsic and mitochondria-dependent intrinsic apoptosis was dependent on the caspases in the chondrocytes treated with 25-HC. Similarly, Olivier et al. reported that 25-HC induced the caspase-dependent apoptosis involving caspase-8 and caspase-3 that was mediated by death receptor-mediated apoptosis in human keratinocytes [5]. Interestingly, they showed that 25-HC induced pyroptosis, a highly inflammatory form of programmed cell death, through the activation of the P2X7 receptor, along with an increase in caspase-1 and pro-inflammatory cytokines [5]. Jang et al. [15] reported that the 25-HC contributed to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the inflammasome. Furthermore, Gold et al. [16] suggested that 25-HC acted as an amplifier of the inflammatory signaling. While all these studies suggest that 25-HC may be closely associated with inflammation, there are also contradictory reports. Reboldi et al. [17] reported that 25-HC suppressed IL-1-driven inflammation and served as a critical mediator in the negative-feedback pathway of interferon signaling in the production of IL-1 family cytokine and inflammasome activity. We also did not observe an increase of inflammation in the chondrocyte treated with 25-HC. Therefore, whether the 25-HC-induced apoptosis is related to 25-HC-induced inflammation in chondrocytes remains to be further investigated.
Interestingly, intra-articular injection of 25-HC into the knee joints not only induced the expression of caspase-3 in the articular cartilage and meniscus, but also increased proteoglycan loss. Recently, Liu et al. [18] reported that 25-HC activated the expression of CH25H in an LXR-dependent mechanism. Hence, the 25-HC-induced proteoglycan loss may be mediated by the expression of CH25H that induces progressive degeneration of ECM through the expression of cartilage degrading enzymes.
Although we demonstrated the pathophysiological role of 25-HC in chondrocytes, there are some limitations to our study. High concentrations of 25-HC were used to investigate apoptosis of chondrocytes in the present study. Although the concentration of 25-HC in the synovial fluid of normal individuals and patients with OA or rheumatoid arthritis (RA) is still not defined, the concentration of 25-HC was reported as approximately 4 ng/ml in the serum of normal individuals [19,20]. Furthermore, the concentration of 25-HC was found to be approximately 11 ng/ml in serum collected 4 h following intravenous injection of small doses (4 ng/kg of body weight) of lipopolysaccharide into healthy male volunteers [20]. Although these studies support that the concentration of 25-HC significantly increases following inflammation, it did not affect the viability of chondrocytes treated with 10 ng/ml 25-HC for 24 h in present study. However, there is a possibility of apoptosis in chondrocytes that are exposed to low concentration of 25-HC for long periods, because the degeneration of articular cartilage progresses over a long-time accompanied with a low-grade inflammation. Hence, it is necessary to determine the physiological concentration of 25-HC in serum and synovial fluid collected from normal individuals and patients with OA or RA, for it to be used as an arthritis-related biomarker. Furthermore, it is necessary to comparatively investigate the apoptosis of chondrocytes and its physiological characteristics following long-term exposure to physiological concentration of 25-HC that is clinically verified from the serum or synovial fluid from normal individuals, and patients with OA or RA.
In conclusion, we demonstrate that inflammation-induced CH25H-25-HC axis acted as a pathophysiological risk factor to induce OA through both death receptor-mediated extrinsic and mitochondria-dependent intrinsic apoptosis in chondrocytes. Hence, our study suggests that cholesterol metabolism associated with metabolic syndrome is closely associated with the development and progression of OA.
 XML Download
XML Download