

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The myocardium has traditionally been considered a passive target of exogenous signaling such as sympathetic innervation, which modulates its chronotropic, inotropic and lusitropic characteristics. To a certain extent this is indeed the case; environmental input from the autonomic nervous system as well as endocrine tissues influence heart rate, contraction and relaxation in response to stimuli. β1 adrenergic receptors, for example, are expressed on cells of the sinoatrial and atrioventricular (AV) nodes as well as on atrial and ventricular cardiac myocytes, where release of norepinephrine from postganglionic neurons of the sympathetic nervous system increases heart rate and contractility in the classical “fight or flight” response. However, there is a growing body of evidence indicating that the heart itself behaves as a sophisticated paracrine and endocrine organ which can actively alter its secretory phenotype in order to meet its metabolic and energetic demands, as well as recruit other cell types such as immune cells in response to damage or inflammation.
The concept of the heart as an organ with endocrine and paracrine signaling, capable of cross-talk with other cells and tissues, arises from the discovery in the 1970s and 1980s that mammalian atrial and ventricular cardiac myocytes secrete small peptide hormones which lower blood pressure via excretion of excess sodium from the kidneys: atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), respectively.1) These are now commonly considered part of the cardiac natriuretic peptide (cNP) family, where circulating levels of ANP and BNP are increased in several cardiac pathologies such as heart failure (HF) and can be used diagnostically as well as prognostically.2) Since then, numerous signaling factors from the myocardium to other target tissues have been identified, such as endothelin-1 and adrenomedullin.1)
Approaching our understanding of the myocardium as an endocrine organ, which actively participates in bidirectional signaling, both in health and pathology, shifts the classical paradigm of the heart as a pump whose function is altered solely by exogenous factors, to one where the heart possesses a great deal of influence and sophistication in altering the metabolic phenotype of other cell and tissue types. Herein is a summary of current knowledge regarding important molecular players involved in the communication between cardiac and adipose tissues, which is one line of endocrine/paracrine communication that has come to the forefront.
MYOCARDIAL ENERGY DEMANDS AND SUBSTRATE UTILIZATION
The heart necessitates an extraordinary demand for fuel in order to function appropriately. As a result, the adult heart obtains up to 90% of its energy from mitochondrial β-oxidation of long-chain fatty acids (FAs); the remainder of energy is supplied from oxidation of substrates such as glucose, lactate and ketone bodies.3)4) These free FAs (FFAs) are provided in large part by fat stores in the body, as activation of lipolysis in adipocytes via β3AR signaling provides energy to the heart; conversely, the heart possesses the ability to regulate adipocyte metabolism through molecules such as ANP and BNP.5) Per gram, FA oxidation provides the greatest energy yield compared to other substrates; however, the process of lipid catabolism required to convert long-chain FAs into acetyl CoA requires a higher oxygen demand than substrates such as carbohydrates6)—a demand for oxygen which is not always available to the myocardium. In these circumstances, other substrates such as glucose, for which adenosine triphosphate (ATP) production is less oxygen-consuming than ATP from FA catabolism, may be preferred.
One of the most remarkable features of myocardial metabolism, therefore, is its flexibility in response to changes in the availability of substrates, exogenous signals such as during development, or physiological health of cardiac myocytes such as in disease. For example, fetal development occurs in a low-oxygen environment where reliance on FA β-oxidation would not provide enough ATP. Substrate preference in fetal hearts is therefore biased toward glucose oxidation for which oxygen demands are lower and more energetically efficient, an observation which has been established for several decades.7)8) The increased availability of oxygen in the postnatal environment initiates a metabolic “switch” from fetal to adult gene transcription which controls substrate utilization, mitochondrial biogenesis, and an adult cellular phenotype in cardiac myocytes. This postnatal gene reprogramming is driven by nuclear receptor peroxisome proliferator-activated receptor (PPAR) α and PPAR-γ coactivator 1 (PGC-1α) wherein PPAR-α is activated by ligand binding of FAs and positively regulates the expression of multiple genes involved in FA uptake and oxidation in cardiac myocytes.9) Additionally, PGC-1α activates PPAR-α, which in turn increases mitochondrial biogenesis and an increase in carnitine palmitoyl transferase I expression and related genes involved in FA substrate utilization.10)
Following ischemic events such as observed during myocardial infarction (MI), which reduce levels of available oxygen, or other cardiac stressors like pressure or volume overload, hyperglycemia or localized inflammation, the adult heart responds to a wide spectrum of stressors by reverting back to a glucose-biased phenotype via fetal gene reprogramming.11) The upregulation of transcription factors such as NFATc1, NKX2-5, GATA4, and MEF2, as well as signaling proteins such as ANP and transforming growth factor β, are responsible for the reverse metabolic switch to fetal substrate utilization,12) wherein changes in metabolic signaling occur prior to pathological changes to the myocardium as seen in hypertrophy and failure. Of note, these include changes in expression of several important metabolic regulators which function in fetal physiology but are primarily pathological in nature when reiterated in the adult heart.
During the transition from non-failing function to HF in the adult human myocardium, for example, levels of glucose transporters such as GLUT1 and GLUT4, pyruvate dehydrogenase kinase 2, medium chain acyl-CoA dehydrogenase and acetyl-CoA carboxylase decrease to those observed in fetal hearts,13) which acutely may rely on the efficiency of glucose oxidation during periods of oxygen unavailability. To further support the concept of metabolic substrate switching acting as an immediate protective mechanism, it has been shown that during hypoxia, increased cardiac glycogen stores in the heart are positively correlated with survival.14)15)16) Conversion of circulating (intracellular) glucose into glycogen during cardiac stress prevents aberrant or excessive glycosylation of proteins in cardiac myocytes as well as oxidative stress induced by glucotoxicity. Additionally, as switching from metabolism of FAs to glucose appears to be a necessary prerequisite for immediate cell survival, increased glycogen stores offer a significant source of energy production during periods of limited substrate availability such as that seen during cardiac ischemia.17) Unsurprisingly, hypoxic events in the heart trigger a rapid downregulation of genes under the control of PPAR-α, which further supports the observation of metabolic substrate switching from FAs to glucose.13)
ADIPOSE TISSUE AND ENDOCRINE SIGNALS TO THE HEART
Adipose tissue can be classified by its anatomical location as well as its phenotype and embryonic origin (Figure 1). Subcutaneous adipose tissue (SAT) can be found immediately below the skin and associated vasculature thoracically and abdominally as well as in the extremities, whereas visceral adipose tissue (VAT) is mainly restricted to the regions surrounding the organs of the torso such as the stomach, liver, kidneys and gastrointestinal tract18) and is associated with hepatic portal drainage. SAT differs from VAT in a number of important features; namely, VAT is more highly vascularized and innervated than SCAT and contains more immune cells such as macrophages. Metabolically, the two adipocyte deposits differ as well; VAT contains higher densities of glucocorticoid and androgen receptors than SAT and as a result, adipocytes comprising VAT are more metabolically active, more insulin‐resistant and more sensitive to lipolysis.19)20) VAT additionally possesses reduced preadipocyte differentiating capacity relative to SAT. Importantly, like epicardial adipose tissue (EpAT), the specialized fat deposit covering the myocardium, VAT has a greater capacity to release FFAs into circulation as well as to uptake glucose than SAT and is more sensitive to adrenergic stimulation.21) SAT, conversely, readily takes up circulating FFAs and triglycerides from the bloodstream. Due to these differences in metabolism and signaling, VAT volume and visceral fat mass are a better predictor of patient mortality than SAT.18)
Figure 1
Metabolic crosstalk is dictated by various tissue types. Adipose tissue deposits are classified by anatomical location and phenotype. Inset: key factors are secreted by both myocardial and adipose tissue, which regulate the functions of each other in both health and pathology.
ANF = atrial natriuretic factor; ANP = atrial natriuretic peptide; BCAA = branched-chain amino acid; BNP = brain natriuretic peptide; miR = microRNA.
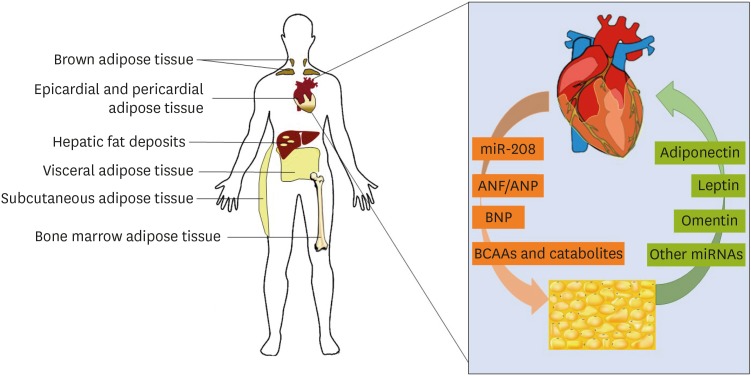
Within non-cardiac AT deposits, adipocytes are further differentiated into white adipose tissue (WAT) and brown adipose tissue (BAT). BAT adipocytes are smaller than WAT adipocytes and, due to the high concentration of mitochondria and extensive vascularization, appear brown in color. BAT mitochondria express high levels of uncoupling protein 1 (UCP1), which is responsible for their greater thermogenic capacity than WAT. This difference in phenotype is partially due to differences in evolutionary origin, as BAT adipocytes are derived from the same origins as skeletal muscle.22) BAT adipocytes also contain a greater number of lipid droplets, which are smaller in size than the single large triglyceride droplet possessed by WAT adipocytes. Of note, WAT adipocytes possess fewer mitochondria than their BAT counterparts, although WAT adipocytes require large amounts of ATP to drive cellular functions such as mitochondrial biogenesis, generation of triglycerides during lipogenesis, release of FFAs into circulation during lipolysis.23) Conversely, BAT adipocytes are of critical importance in neonates as well as during cold stress, and rather than releasing stored triglycerides as circulating FFAs for use by other tissues, FA oxidation in BAT adipocytes drives thermogenesis.24) Within WAT deposits exist adipocytes which are derived from WAT but possess with some characteristics of brown adipocytes. First described several decades ago in mouse models25) these cells are now referred to as beige or brite (brown + white) adipocytes. Beige adipocytes phenotypically resemble brown adipocytes due to the presence of multilocular lipid droplets, greater number of mitochondria, and high UCP1 expression,26) and WAT can be induced to “browning” by cNPs or by PGC1-α expression and irisin secretion from the skeletal muscle27) as examples of cardiokine-mediated crosstalk in mouse models.28)
EPICARDIAL ADIPOSE TISSUE AND LOCALIZED HEART-FAT CROSSTALK
EpAT is located between the visceral pericardium and the heart, in direct contact with the myocardium derived from the splanchnopleuric mesoderm which is developmentally associated with the digestive tract.29) Possessing features of both WAT and BAT, EpAT primarily lies along the AV and interventricular grooves, as well as over the free wall of the right ventricle and apex of the left ventricle, where the coronary arteries provide its blood supply.30) This location allows EpAT to regulate cardiac metabolism via secretion of molecules such as adiponectin,31) as well as receive signals from the heart in response to injury or disease.32) EpAT is a metabolically active organ which generates FFAs and inflammatory cytokines, and as such, recent studies suggest that this localized fat distribution plays an important role in the development of pathology such as HF, type II diabetes mellitus and metabolic syndrome.33) Currently, EpAT thickness is utilized as a clinical biomarker correlated to features of both HF and metabolic syndrome such as left ventricular (LV) mass, mean arterial blood pressure, and circulating low-density lipoprotein cholesterol, fasting insulin, and adiponectin levels.34)35) Due to its proximity and role in cardiac crosstalk, EpAT is highly specialized in its ability to provide energy and maintain energetic homeostasis in the heart. Compared to visceral and subcutaneous fat deposits, EpAT releases and takes up circulating FFAs at a much higher rate, and conversely utilizes glucose at much lower levels, and as FFAs serve as the primary energy substrate for cardiac myocytes, these high FA metabolic rates may serve as a buffer against lipotoxicity while enabling rapid access to preferred cardiac substrate,36) much in the same way uptake of glucose into glycogen buffers hepatocytes against glucotoxicity and glycation of proteins.10)
ADIPOKINES AND CARDIOKINES: KEY MESSENGERS BETWEEN THE HEART AND FAT
Leptin
The first signaling molecule discovered which is secreted by adipocytes was the small peptide hormone leptin in 1994.37) Leptin acts predominantly on receptors in the hypothalamus to modulate hunger and inhibit lipid storage in adipocytes,37) where its loss has been linked to obesity and skeletal muscle remodeling in both human subjects and in animal models such as in leptin-deficient ob/ob mice.38) Additionally, obese patients demonstrate high circulating levels of leptin due to an increased fat mass39) and leptin insensitivity, similar to insulin resistance observed in patients with type 2 diabetes, diminishes satiety signaling which in turn exacerbates HF.40) Since then it has been shown that separate from its role in modulating obesity which can indirectly drive HF, leptin can also act directly on the myocardium41) where it performs several functions including FA and glucose metabolism and protection against apoptosis during stress. The human heart highly expresses leptin receptors, and while it has been demonstrated that leptin is sufficient to trigger growth of cultured cardiac myocytes,41) this claim is controversial as not all studies demonstrate hypertrophic effects of leptin in vitro. Interestingly, clinical studies demonstrate an association of circulating plasma leptin levels with cardiac hypertrophy; specifically, several studies involving insulin resistant patients showed a positive correlation between fasting plasma leptin levels with myocardial wall thickness, but not with LV mass, even after correcting for body mass index, which suggests that leptin plays a role in LV hypertrophy.42) Of note, cardiac myocytes isolated from ob/ob mice demonstrate diminished expression of the leptin receptor, hypertrophy, and contractile defects such as decreased peak shortening and lower maximal velocity of myocyte shortening and relengthening; these defects were not observed in age- and gender-matched high fat-induced obese counterparts with normal leptin signaling.43) During cardiac injury, leptin appears to play a protective role; for example, cardiac ischemia has been reported to have varying effects on expression of leptin receptors, with ischemia/reperfusion studies in isolated Sprague-Dawley rat heats demonstrating that ischemic injury was associated with decreased leptin receptor expression.44)
Adiponectin
Adiponectin, also referred to as GBP-28, apM1, and AdipoQ, is a small (<250 aa) polypeptide hormone in the complement 1q family and encoded by the ADIPOQ gene.45) Produced by adipocytes. adiponectin belongs to the complement 1q family but bears structural homology to tumor necrosis factor alpha (TNF-α).46) The primary functions of adiponectin are regulation of glucose levels and catabolism of FFAs through β-oxidation47); as a result, dysregulation of adiponectin levels has been implicated in various metabolic pathologies. Obese individuals have been reported to possess lower circulating adiponectin levels compared to non-obese individuals,48) as well as in patients with type II diabetes mellitus and metabolic syndrome.45) Conversely, its overexpression suppresses adipocyte growth and differentiation49) with marked increase in energy expenditure and upregulation of uncoupling proteins.
Given that obesity is a well-established risk factor for pathologic cardiac remodeling and hypertrophy,50) and that insulin resistance manifests as diastolic dysfunction even early in its development,51) studies in the past decade have shown that adiponectin is a key player in the development of pathological cardiac remodeling during HF in large part due to its role in insulin resistance. Mirroring what is observed in patients with type 2 diabetes mellitus, streptozotocin-induced diabetic rats demonstrate markedly reduced circulating plasma adiponectin levels but a cardiac-specific upregulation of adiponectin receptor 1 along with decreased phosphorylation of adenosine monophosphate-activated protein kinase and reduced expression of the glucose transporter GLUT4.52) Cardiac glucose utilization is dependent on availability of and sensitivity to insulin, and decreased responsiveness of the heart to metabolic regulation such as provided by insulin and adiponectin signaling, can create a state of cardiac metabolic dysfunction which is a risk factor for the development of cardiovascular diseases (CVDs) such as hypertension-induced hypertrophy.53)54) Further linking reduced adiponectin levels and insulin resistance, adiponectin-knockout murine models demonstrate slower plasma clearance of FFAs, reduced FA transport protein 1 levels, and increased TNF-α levels in adipocytes as well as plasma.55) The effects of adiponectin loss in adipocytes triggers molecular changes in myocytes such as severely reduced insulin-receptor substrate 1 (IRS-1)-associated P13K activity.
In the context of acute myocardial damage, such as seen in infarction, adiponectin plays a critical role in myocardial wound healing, as circulating adiponectin not only directly signals to cardiac myocytes, but enhances the secretion of the anti-inflammatory cytokine interleukin-10 by macrophages and in fact promotes macrophage polarization toward the anti-inflammatory M2 phenotype required for wound healing post myocardial damage.56) Adiponectin signaling from adipose tissue to the myocardium, additionally, exerts anti-apoptotic and antioxidant effects on cardiac myocytes following damage to the heart via activation of the enzyme ceramidase which is associated with the adiponectin receptors AdipoR1 and AdipoR2.57) Of importance, specific myocardial injury such as seen in patients undergoing heart surgery, which induces acute oxidative stress, triggers compensatory upregulation of adiponectin levels via cardiac myocyte-derived release of lipid peroxidation products which signal to adipocytes in EpAT to stimulate PPAR-γ activity and increase production of adiponectin from the epicardial fat pad to the injured myocardium.31) This example of finely-tuned bidirectional crosstalk between adipocytes and cardiac myocytes highlights the importance of fat in cardiac health as well as pathology, particularly as myocardial oxidative stress is an important predictor of the development of cardiac diseases and the loss of adipokine signaling contributes to not only the development of chronic cardiac disease but acute pathology such as atrial fibrillation observed in patients following heart surgery.31)
Omentin
Also known as intelectin-1, this peptide is a recently identified novel adipokine which is still poorly characterized; however, it is expressed primarily in EpAT and omental adipocytes58) and is implicated in directed crosstalk with the myocardium and may be a major player in the protective effects of the EpAT following myocardial injury. Of note, obese patients and animal models report reduced circulating plasma levels and RNA-level expression of omentin,59) and independent of obesity similar reduction in circulating omentin levels are observed in patients with metabolic syndrome and type 2 diabetes.60) Reduced circulating levels of omentin are positively correlated with CVD such as atherosclerosis, HF, and MI.61) It has been shown experimentally in animal models that omentin has protective effects against myocardial ischemia-reperfusion damage62) and that during ischemic injury, omentin expression decreases myocardial hypertrophy and is pro-angiogenic.60) Additionally, omentin treatment causes vasodilation via endothelium-dependent nitric oxide signaling in isolated rat aorta63)64) and inhibits vasoconstriction following treatment with noradrenaline.
Branched-chain amino acids
Named due to the structure of the side chain possessing a branched carbon moiety, branched-chain amino acids (BCAAs) are comprised of the essential amino acids valine, leucine and isoleucine and are regulated by catalysis of proteins in cells such as myocytes, hepatocytes and adipocytes, and their homeostasis is determined largely by catabolic activities in a number of organs including liver, muscle and adipose tissue.65) It is known that elevated circulating plasma levels of leucine, isoleucine and valine are frequently found in, and positively correlated with, increased CVD risk, and that dietary BCAA intake, particularly for leucine, is linked to increased diagnosis of CVD such as atherosclerosis.66) Recent genomics studies have identified the transcription factor Krüppel-like factor 15 as a key upstream regulator of cardiac BCAA catabolism, and that defects in normal BCAA catabolism promoted pressure overload-induced HF67) and increased mitochondrial oxidative stress in cardiac myocytes.
Excitingly, recent studies of cardiac G protein-coupled receptor kinase (GRK) activity have unveiled a previously unknown level of whole-body metabolic control, namely adiposity, by the heart. GRK2 and its regulated activity in cardiac myocytes specifically has been identified as a systemic metabolic modulator; for example, it has been shown that GRK2 can promote insulin resistance in the heart by phosphorylating IRS-1.68) Woodall et al.69) demonstrated the importance of cardiac control of BCAA metabolism on adipocyte differentiation by comparing the effects of a high-fat diet (HFD) on control mice compared to cardiac-specific transgenic mice expressing a C-terminus peptide of GRK2 known as the βARKct that is an inhibitor of GRK2 activity (TgβARKct). TgβARKct mice demonstrated a significant increase in several metabolites belonging to the BCAA pathway both in heart tissue as well as in serum circulation, including α-hydroxyisovalerate (HIVA) and 2-hydroxy-3-methylvalerate (HMVA). Interestingly, TgβARKct mice that have decreased GRK2 activity, gained significantly more weight than non-transgenic control mice after a HFD and this was manifested in significantly more visceral fat in number and size of adiposites.69) Conversely, transgenic mice with cardiac-specific overexpression of GRK2 (TgGRK2) demonstrated opposite changes in BCAA intermediates to those seen in TgβARKct mice fed a HFD and also reciprocally, TgGRK2 mice had a lean phenotype after HFD compared to their non-transgenic controls.69) As it has been established that elevated BCAA concentrations have been linked to obesity, insulin resistance and metabolic syndrome, these experiments more closely link cardiac-secreted factors with peripheral adiopogenesis as circulating metabolites differentially regulated by GRK2 signaling in the heart was acting as a cardiac-generated signaling molecule. Due to the reciprocal nature of the obesity phenotype after a HFD in mice with gain or loss of myocyte GRK2 activity along with reciprocal cardiac and circulating BCAA catabolites, several α-hydroxy BCAA metabolites, HIVA, HMVA, and α-hydroxyisocaproate, were examined as potential adipogenic cardiokines. It was discovered that HMVA demonstrated a positive effect on lipid accumulation in differentiated 3T3-L1 cells which was dose-dependent69); this suggests that changes in BCAA metabolism in the heart trigger altered release of cardiokines such as HMVA, which are regulators of adipocyte differentiation and thus, behavior of metabolically important cells such as adipocytes. This study also identified other small molecule metabolites reciprocally regulated in TgβARKct and TgGRK2 mice that have the potential to also communicate with systemic adipocytes including endocannabanoids.69) However, the study did not rule out larger protein factors secreted by the myocyte that is responsible for changing adiposity after a HFD when GRK2 levels are changed. In any regards, since GRK2 is up-regulated in the injured or stressed heart,70) changes in GRK2 activity in the myocyte and subsequent regulation of adipocytes is something extremely important to consider in future studies.
Given the importance of protein turnover in healthy cardiac function and the correlation between defective BCAA metabolism and CVD,71)72)73) it is also important to acknowledge the critical role adipocytes play in regulating AA metabolism. It is known that BCAA catabolism promotes adipogenesis74) and as demonstrated above, this mechanism is partially under cardiac crosstalk control. Additionally, it is known that a metabolic substrate switch occurs between proliferating cells, which use glucose and glutamine to generate acetyl-coenzyme A for tricarboxylic acid (TCA) cycle entry, and differentiated adipocytes which prefer BCAAs such as leucine and isoleucine and which contribute as much as a third of acetyl-coenzyme A source substrate.75) Recently, whole-body isotopic tracing of BCAAs in healthy and insulin-resistant mice76) in order to determine their tissue-specific fate for metabolic oxidation has uncovered that under normal conditions, the myocardium, skeletal muscle, BAT, and liver utilize the greatest amount of BCAAs for TCA cycle substrate; however, insulin-resistant mice shifted BCAA usage in myoyctes and demonstrated reduced BCAA oxidation in adipocytes and liver, likely affecting cardiac metabolism under these conditions.
Atrial natriuretic peptide and brain natriuretic peptide
Natriuretic peptides were the first cardiokines discovered, with ANP or factor identified as a 28-amino acid molecule secreted by granules in the atria77) which triggered diuresis from rat kidney when purified and administered exogenously.78) BNP was subsequently discovered in atria from a porcine model and confirmed in humans79)80) as having similar effects as ANP. Both proteins are released by cardiac myocytes in response to hypervolemic states, stimulation with angiotensin II stimulation and endothelin, and β-AR activation.81) Elevated levels of ANP and BNP are found during HF, and circulating levels of BNP and its larger precursor protein NT-pro-BNP are sensitive, diagnostic markers utilized clinically to assess HF in patients.81) Aside from their effects in modulating the effects of the renin-angiotensin-aldosterone system, ANP and BNP are now known to signal to adipose tissue.5) Plasma levels of both ANP and BNP are reduced in people with obesity, type 2 diabetes mellitus and metabolic syndrome,82) and cardiac mRNA expression of both ANP and BNP in db/db mice83) and obese Zucker rats84) is significantly lower than healthy controls. Overall this indicates that metabolic regulation by natriuretic peptides is inversely correlated with overall cardiovascular risk. Importantly, adipocytes possess receptors for both ANP and BNP,85)86) where they have such effects as increasing lipolysis via an adenylyl cyclase-independent and cGMP-dependent mechanism and modulate the release of adipokines.87) Therefore, altered cardiac secretion of ANP and BNP may be potentially linked to obesity and metabolic syndromes, rather than a consequence of adipokine dysregulation affecting cardiac health.
MicroRNAs
MicroRNAs (miRs) are small, highly-conserved noncoding RNAs involved in post-transcriptional gene regulation and protein translation. Currently over 2,000 miRs have been identified.88) Their role in both development and health as well as pathology indicate a potential involvement in cardiac-adipose crosstalk since they are abundantly found in the circulation. One of the first identified miRs, a 20–24 nucleotide noncoding RNA transcript, was determined to be myocyte specific with expression detected by embryonic day 8.5 in murine hearts and an increase in expression throughout cardiac development.89) The miR-27 family is indirectly implicated in HF due to its role in cholesterol homeostasis, FA metabolism, and adipocyte differentiation.90) Levels of miR-27 are downregulated during adipocyte differentiation; conversely overexpression of this miR family inhibits adipocyte formation when overexpressed, primarily through post-transcriptional inhibition of by miR-27a and -27b as well as inhibition of C/EBP alpha in by miR-27b.91)
The effects of miRNA regulation on adipocyte crosstalk with the heart are mostly indirect and involve miR-mediated lipogenesis, browning or inflammation and the resultant endocrine signals released by adipose tissue. As a result, these miRs can be classified as pro-adipogenic or anti-adipogenic in nature.92) Examples of miRs which promote adipocyte differentiation include miR-143, miR-103, miR-146b, miR-148, and miR-33b; their upregulation promotes activity of PPARγ2, cell cycle transcription factors, FABP4, GLUT4, and adiponectin and levels of pro-adipogenic miRs are inversely correlated with type 2 diabetes, obesity and other CVD risk factors.92)93) Conversely, anti-adipogenic miRs include miR34a, miR-125-5p and miR-200b/a. MiR-34a for example, is released from adipocytes and circulating levels are increased in serum from diabetic patients.94) It is now known that circulating miRs are released from AT in exosomes, and confer endocrine-like effects on other tissues.95) The implications of miR-based crosstalk between the heart and fat, and its effects on myocardial health demand further study, particularly as differences in circulating miR populations exist in patients with CVD risk factors. A large clinical study comparing circulating miRs of obese vs lean children uncovered a cohort of differentially-expressed miRs; miR-486-5p, miR-486-3p, miR-142-3p, miR-130b, and miR-423-5p were upregulated and correlated positively with insulin resistance, high-molecular-weight adiponectin, C-reactive protein levels, and circulating FFAs.96)
A small number of consistently stress-responsive dysregulated miRs in CVD have emerged from these studies, however, with postmortem comparison of cardiac tissue from patients who experienced MI to that of trauma victims without cardiac disease and fetuses without cardiac abnormalities revealed upregulation of miR-208 and downregulation of miR-1 and miR-133a from infarcted cardiac tissue compared to healthy adult and fetal hearts.97) Another pathological cardiac miR identified recently is miR-195, which in human hearts is consistently upregulated during pathological hypertrophy, and murine models of cardiac-specific miR-195 overexpression results in dilated cardiomyopathy and decompensatory HF in mice.98) Mir208 in particular has become an attractive target for HF studies. The miR-208 family consists of miR-208a and miR-208b, which are highly homologous in sequence; however, miR-208a is encoded by an intron of Myh6 whereas miR-208b is located within an intron of Myh7.99) Myh7 is predominantly expressed in the embryonic hearts of mice and the majority of myosin isoform expression transitions to Myh6 postnatally, which mirrors a switch from miR-208b to miR-208a expression during birth.100) The role of the miR-208 family in driving HF is highlighted in the response of miR-208–/– mice to cardiac stressors; following pressure overload by thoracic aortic constriction or signaling by calcineurin mice lacking miR-208–/– were resistant to cardiac hypertrophy and fibrosis, did not experience upregulation of myosin heavy-chain beta (the product of MYH7) as observed in wild type (WT) mice.12)
However, the mechanisms responsible for the protective effects of miR-208 deletion were not elucidated until recently. Following the observation that MiR-208a−/− hearts resembled hearts of hyperthyroid patients, which are, interestingly, also protected against pathological hypertrophy and fibrosis. Grueter et al.101) determined that one of the validated targets of miR-208a is a component of the Mediator complex, a multi-protein machine which modulates thyroid hormone-dependent transcription, named MED13. First identified in a Drosophila model as a negative regulator of lipid accumulation, knockdown of MED13 using siRNA targeted with a muscle-specific promoter caused excessive adiposity,102) pharmacological studies utilizing mice treated with a specific miR-208a inhibitor (anti-miR-208a) over several weeks were leaner than control littermates without changes to no effect on cardiac size, weight or cardiac contractility. Similar findings were observed in transgenic mice overexpressing MED13 specifically in the heart (MED13cTg, mice), which were lighter and possessed less fat mass with smaller adipocytes than WT littermates and demonstrated resistance to obesity when fed a HF diet. Interestingly, both visceral WAT and subscapular BAT mass were reduced Med13Tg mice on a HFD. Given that a reverse phenotype occurs with cardiac deletion of MED13, including increased susceptibility to diet-induced obesity and metabolic syndrome, miR208 has emerged as a critical modulator of whole-body metabolism and energy homeostasis. More recent studies from this group103) have further established a role for miR-208 in cardiac regulation of target tissues by showing that the lean phenotype observed in MED13cTg mice is correlated with increased lipid uptake, FA β-oxidation, and total mitochondrial content in both WAT and liver. Most importantly, the cardiac overexpression of MED13 seen in MED13cTg mice decreases the expression metabolic genes in the heart, but upregulates these genes in WAT,103) and parabiosis of MED13cTg and WT mice demonstrate that circulating factors in MED13cTg mice confer enhanced metabolism and a lean phenotype to attached WT mice. Importantly, studies in our lab, ruled out this pathway contributing to the altered post-HFD obesity phenotype in cardiac-specific GRK2 altered mice,69) demonstrating that GRK2 activity is regulating novel signaling leading to release of cardiokines that can regulate fat.
CONCLUSION
As cardiovascular research increasingly supports the concept of the myocardium as a dynamic, communicative organ with the ability to secrete a wide range of messengers known as cardiokines, and adipose tissue as an important metabolic tissue whose adipokines relay messages to the heart, it is critical to assess current knowledge of crosstalk between these tissues. The nature of these signals is truly bidirectional, as damage to the myocardium has been shown to impact peripheral metabolic changes; conversely, systemic diseases such as obesity and type 2 diabetes mellitus are associated with a number of direct phenotypic changes to the myocardium as well as indirect effects like inflammatory cell recruitment which in turn exacerbate CVD. As new members of the heart-fat secretomes are identified, there exists an increasing emphasis on the importance of beneficial molecular messengers such as adiponectin on myocardial health, and not simply detrimental biomarkers released from AT. Additionally, further research is needed to examine the unique and double-edged nature of EpAT, a truly unique crosstalk player which can impact cardiac health both protectively and during pathology. Ultimately, there exists a delicate and dynamic balance between adipokines and cardiokines that determines the consequences of metabolic disease and CVD on one another. Most importantly, little is yet known about the balance of protective cytokines to and from the heart in the context of pathology, where such novel cardiokines offer both therapeutic targets and clinically relevant biomarkers. The heart will continue to reveal itself as a highly sophisticated endocrine and paracrine organ, capable of speaking many languages as it negotiates supply and demand with a complex landscape of adipose tissues as well as the rest of the body.
 XML Download
XML Download