

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Staphylococcus schleiferi subsp. schleiferi (S. schleiferi) was initially described in 1988 and has been reported to be associated with a number of infections in companion animals and humans [1]. Although coagulase-positive Staphylococcus pseudintermedius has most frequently caused canine otitis externa and skin and soft tissue infections [2], recent studies reported that coagulase-negative S. schleiferi is becoming more prevalent in canine otitis externa and pyoderma cases [234]. Furthermore, the increased frequency of methicillin-resistance in S. schleiferi (MRSS) is of significant public health concern worldwide [456]. A recent study in our laboratory has indicated that ~26% of S. schleiferi strains of the canine origin were MRSS, often presenting increased levels of multiple drug resistance phenotype [6]. Genotypic characterization of these canine-associated MRSS strains revealed that the methicillin resistance was frequently conferred by staphylococcal cassette chromosome mec type V (SCCmec V) [6]. The widespread occurrence of SCCmec V has been reported in other methicillin-resistance staphylococci, in particular, in livestock-associated methicillin-resistant S. aureus (MRSA) [7], indicating dissemination of the SCCmec V among various staphylococcal species. Although MRSS strains have been increasingly isolated from the infected dogs over the past decades worldwide [34], complete whole-genome sequences are available for only several S. schleiferi strains isolated in the USA and Japan. Thus, a representative canine-associated MRSS strain with SCCmec V isolated from Korea was selected for complete genome sequence analysis in order to understand its prevalence, genetic repertoire, and relationship to the previously sequenced MRSS strains.
MATERIALS AND METHODS
A SCCmec V-MRSS strain designated as SS4 was isolated from an ear swab sample of a dog with otitis externa in Seongnam, Korea, in 2017 [6]. The strain was identified as S. schleiferi using the Vitek 2 system (BioMérieux, France), sequencing of 16S rRNA and tuf gene (Cosmogenetech, Korea), and coagulase production test. This strain was resistant to several antimicrobial agents, including ampicillin, enrofloxacin, kanamycin, and oxacillin. Genotypic analyses revealed that this strain possessed SCCmec V and five of the staphylococcal enterotoxin genes (seg, sei, sell, selm, and selq). This strain had oxacillin MIC of 8 μg/mL. The S. schleiferi strain was grown in tryptic soy broth (TSB, Difco Laboratories).
Genomic DNA (gDNA) of S. schleiferi SS4 strain was extracted using the Wizard genomic DNA Isolation Kit (Promega, USA), according to the manufacturer's protocols. Quantity and quality of the extracted gDNA was assessed via fluorescence-based quantification and NanoDrop 2000c spectrophotometer (Thermo Scientific, USA), respectively.
Whole genome sequence data of S. schleiferi SS4 strain were generated by a combination of PacBio RS II (Pacific Biosciences, USA) and Illumina HiSeq platform (Illumina, USA). Library preparation of de novo genome assembly was carried out by PacBio reads under the Hierarchical Genome Assembly protocol (version 3.0) in SMRT Portal version (2.3.0). PacBio sequencing yielded 136,862 reads covering 1,305,140,901 bp with genome coverage of 514X. After the de novo assembly, Illumina HiSeq generating 150 bp paired-end reads was applied for rectifying the error using the Pilon program (version 1.21). Following the hybrid assembly, 5,277,928 reads covering 796,967,128 bp with 313X coverage were generated from Illumina sequencing. Genome annotation was performed using Prokka 1.12b.
For comparative genomic analyses, the previously published genome sequences of five S. schleiferi strains were obtained from NCBI. The five S. schleiferi strains were all isolated from canine skin infections (accession numbers of the five S. schleiferi strains were 1360-13; CP009470, 2142-05; CP009762, 5909-02; CP009676, 2317-03; CP010309 and TSCC54; AP014944, respectively) [89].
Functional genome analysis of S. schleiferi strains was performed via genome-wide approach, such as proteomics and metabolomics. Gene annotation based on orthology and functionality was done by Cluster of Orthologous Groups and RAST server of SEED databases (http://rast.theseed.org/FIG/rast.cgi) [10]. The presence of virulence genes and antimicrobial resistance genes was confirmed by using BLAST algorithm and Center for Genomic Epidemiology (CGE) (http://www.genomicepidemiology.org/). For identification of antimicrobial resistance genes, ResFinder of CGE and the Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/) were integrated. Mobile genetic elements (MGEs), such as insertion sequences (ISs) and prophages, were detected by using the IS Finder database (https://isfinder.biotoul.fr/) and PHAST (http://phast.wishartlab.com/), respectively.
Phylogenetic analyses of S. schleiferi strains relied on two comparative parameters: average nucleotide identity (ANI) values and 16S rRNA gene sequencing. The ANI values were calculated based on BLAST algorithm (ANIb) using JSpecies [11], and modified ANI was calculated using OrthoANI (version 0.93) [12]. A phylogenic tree of 16S rRNA sequences was generated using the MEGA-X software, and these sequences were aligned with ClustalW. The phylogenic tree was constructed using maximum likelihood analysis implemented with general time reversible model. Pan-genomic analysis of S. schleiferi strains was also performed using the Roary [13].
RESULTS AND DISCUSSION
The complete genome of MRSS SS4 strain comprised a single circular chromosome of 2,539,409 bp with a guanine-cytosine (GC) content of 35.90% and no plasmid. We identified 2310 open reading frames (ORFs), 59 tRNAs, and 16 rRNAs in this genome. The genome size and GC content of the SS4 were similar to those of the five S. schleiferi strains sequenced previously (Supplementary Table 1).
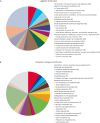
As presented in Fig. 1A and B, the functional categorization of the SS4 genome revealed that categories E (amino acid transport and metabolism), P (inorganic ion transport and metabolism), and J (translation, ribosomal structure and biogenesis) were most abundant (Fig. 1A). SEED data also revealed that 1708 ORFs (70%) encode proteins with known functions, whereas 741 ORFs (30%) produce proteins with unknown functions (Fig. 1B). Among the predicted ORFs, genes involved in the metabolism of amino acids and their derivatives (256 ORFs) were most abundant, followed by those involved in carbohydrate (213 ORFs) and protein (171 ORFs) metabolism. Interestingly, the number of phages, prophages, and transposable elements was higher in the SS4 strain than the other S. schleiferi strains (data not shown). Correlating with the higher number of MGEs, five different enterotoxin genes (seg, sei, sell, selm, and selq), which is usually carried by phages, were identified in the SS4 strains [6]. Different host factors and environmental conditions affecting staphylococcal/microbial communities on canine hosts may have caused the differences in prevalence of phages among the six S. schleiferi strains.
Fig. 1
Functional categorization of annotated genes in the Staphylococcus schleiferi SS4 strain using (A) Cluster of Orthologous Group and (B) SEED databases.
In accordance with the previous publication [6], the SS4 strain harbored mecA gene within SCCmec type V. Although the phenotypic antimicrobial susceptibility profiles of the five S. schleiferi strains were unavailable, three of the five S. schleiferi strains (2317-03, 5909-02, and TSCC54) also had mecA in their genome. Notably, the 2317-03 and TSCC54 strains isolated from dogs in the USA and Japan, respectively, carried SCCmec V, and 5909-2 strain from the USA had SCCmec IVa for methicillin resistance. Analyses of antimicrobial resistant genes in the SS4 and five S. schleiferi genomes revealed that SS4 and 2713-03 strains carried two aminoglycoside resistant gene cassettes, AAC(6′)-Ie-APH(2′′)-Ia and ANT(4′)-Ib. Furthermore, another beta-lactam resistant gene, blaZ, was identified only in the 2317-03 strain (Supplementary Table 2). The three antimicrobial resistance genes identified in the SS4 strain (Table 1) were all located within MGEs.
Table 1
Antimicrobial resistance genes and virulence genes of Staphylococcus schleiferi SS4 strain

CGE and genome BLAST search revealed that the MRSS strain SS4 comprises various virulence factors associated with its potential pathogenicity. As presented in Table 1, three heavy metal resistance genes, eight adhesion-associated genes, four toxin genes, and two exoenzyme genes were identified in the SS4 strain. Of these virulence-related genes, multiple copies of sdrD, hldB, lukS, and lip genes were identified in the SS4 strain. Sequence analysis around the sdrD, hldB, lukS, and lip genes revealed that there are no MGEs or characteristics of translocatable sequences, suggesting that homologous recombination may have caused the amplification of the genes.
Phylogenomic analysis of S. schleiferi strains based on 16S rRNA sequences revealed that all the six S. schleiferi strains were genetically identical (data not shown). In the Roary analysis, S. schleiferi strain SS4 exhibited a close relationship to the 5909-02 strain isolated from infected dogs in the USA (Fig. 2A). Correlating with the Roaryanalysis result, OrthoANI values also indicated that SS4, 2142-05, and 5909-02 are closely related. These data suggested that the MRSS SS4 strain is more closely related to the S. schleiferi strains isolated from the USA than to the TSCC54 strain isolated from Japan. Multiple genome alignment of the six S. schleiferi strains using Mauve algorithm [14] also identified two distinct blocks of genome only in SS4 and 2317-03 strains comprising aminoglycoside resistant gene cassette and cell division-associated proteins, respectively (data not shown).
Fig. 2
Phylogenetic analysis of six genomes of Staphylococcus schleiferi strains based on the pangenomes with Roary (A) and OrthoANI (B).
*Methicillin-resistant S. schleiferi strains: TSCC54, SS4, and 2317-03 had SCCmec V and 5909-02 strain had SCCmec IVa.

To our knowledge, this is the first report of complete genome sequence of an MRSS-SCCmec V strain isolated from a dog with otitis externa in Korea. This whole genome sequence information will contribute to the understanding of genetic features of canine-associated MRSS strains, such as antimicrobial resistance and virulence-related genes. More detailed comparative analysis of the S. schleiferi genomes to those of other staphylococcal species, such as MRSA and methicillin-resistant S. pseudintermedius (MRSP) is necessary for future investigation.
The complete genome sequence of methicillin-resistant S. schleiferi strain SS4 has been deposited in GenBank under the accession number CP035007.
 XML Download
XML Download