

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hearing loss (HL) is genetically highly heterogeneous. More than 100 genes have been identified in patients with nonsyndromic HL (NSHL), and more than 400 genes are known to be associated with some type of syndromic HL (SHL) [1]. NSHL accounts for approximately 70% of hereditary HL, and the deafness loci (DFN) involved are named based on their mode of inheritance: DFNA (autosomal dominant, AD), DFNB (autosomal recessive, AR), and DFNX (X-linked).
Most forms of hereditary HL caused by pathogenic variants (PVs) of a specific gene show only one inheritance pattern; however, a few can have either AD or AR mode of inheritance such as those associated with PVs of GJB2, GJB6, MYO7A, and TECTA [1]. USH1C, encoding harmonin, is known to be related to DFNB18, AR retinitis pigmentosa (RP) with late-onset HL, and sector RP and HL, as well as Usher syndrome type 1 [23456]. All these phenotypes are inherited in the AR mode; an AD inheritance pattern has yet to be reported.
We identified a novel USH1C variant in a large Korean family with NSHL. Interestingly, this family showed a typical pattern of AD inheritance and the USH1C variant segregated perfectly with NSHL. Functional analysis of the USH1C variant revealed impaired binding between cdh23 and the PDZ2 domain of harmonin and suggested a novel pathogenic mechanism, in which a USH1C variant caused AD-NSHL.
MATERIALS AND METHODS
Individuals and clinical evaluation
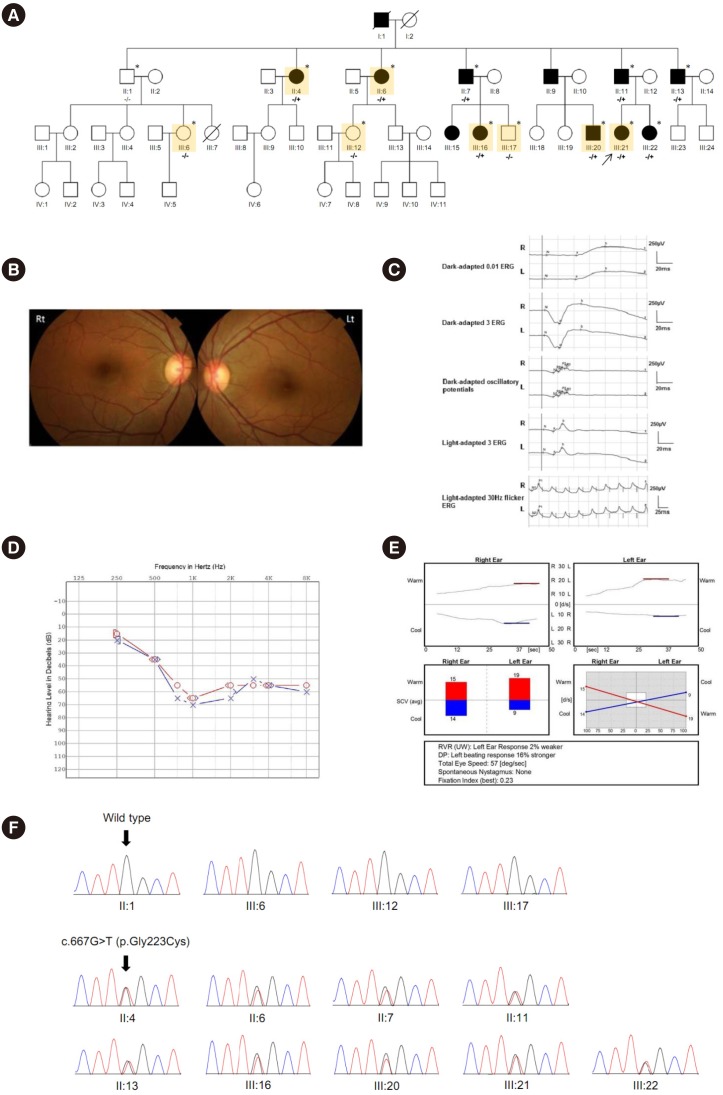
In 2016, a three-generation, 48-member Korean family presenting with AD-NSHL was identified at Samsung Medical Center, Seoul, Korea (Fig. 1A). The proband (III-21) underwent complete ophthalmic examinations, including visual-acuity measurement, slit lamp biomicroscopic examination, fundus examination, color vision test, full-field electroretinogram, color fundus photography, fundus autofluorescence, and spectral domain optical coherence tomography. Audiologic evaluations included pure tone audiometry with air and bone conduction, speech audiometry, impedance audiometry with tympanometry and stapedial reflex, otoacoustic emission, and auditory brainstem response. Vestibular function of the proband was evaluated by caloric test, vestibular evoked myogenic potential, rotatory chair test, and sensory organization test. This study was approved by the Institutional Review Board of Samsung Medical Center, and written informed consent was obtained from all individuals who participated in this study.
Clinical findings
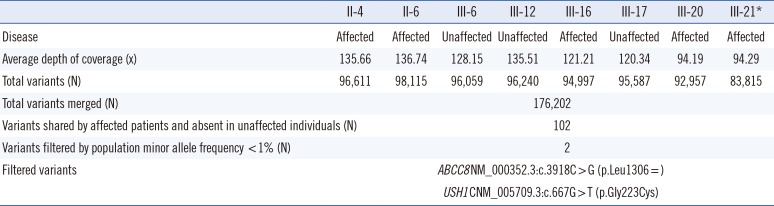
Detailed family history revealed the pedigree of the three-generation, 48-member Korean family with NSHL showing a pattern of AD inheritance (Fig. 1A). For available family members, onset of HL occurred during adolescence in all affected individuals except one (II-13) with prelingual HL and another one (III-16) with later onset in the mid-twenties. Other related clinical manifestations suggesting SHL were not present in any of the affected individuals (Table 1).
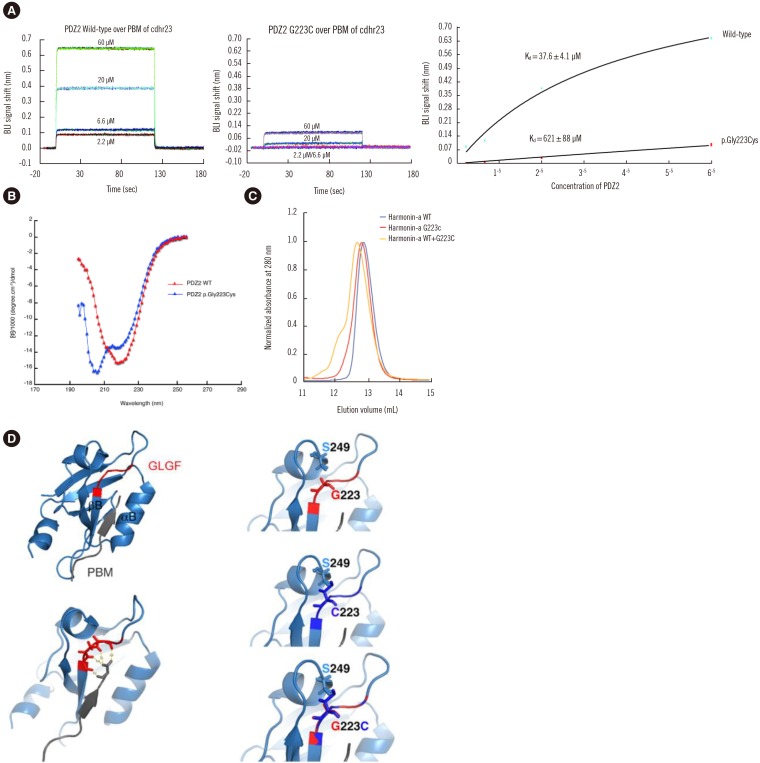
Comprehensive ophthalmologic examinations, including fundus photography and electroretinogram of the proband (III-21), showed normal findings (Fig. 1B and 1C). Pure-tone audiometry demonstrated bilateral sensorineural HL with a pure-tone average (0.5, 1, 2, and 4 kHz) of 53 dB and 56 dB in the right and left ear, respectively (Fig. 1D). The word recognition scores of the right and left ears, presented at the most comfortable level, were 96% and 94%, respectively. Tympanometry indicated a normal type A curve for the tympanogram, and the stapedius reflex (acoustic) was present at all sound frequencies. Distortion product and transient evoked otoacoustic emissions were absent on both sides. The thresholds obtained by auditory brainstem response recordings were 60 dB for both ears. The caloric test, vestibular evoked myogenic potential, rotatory chair test, and sensory organization test showed normal or unremarkable findings (Fig. 1E).
Whole exome sequencing (WES)
To identify a causative gene in this family, we performed WES using genomic DNA extracted from peripheral blood leukocytes. In brief, 3 mL of whole blood was drawn from each individual, and 200 µL of fresh whole blood was used to extract genomic DNA using MagNA Pure 96 Instrument (Roche Diagnostics, Basel, Switzerland). Five affected (II:4, II:6, III:16, III:20, and III:21) and three unaffected individuals (III:6, III:12, and III:17) were selected for WES considering the distance of relatedness [7]. Library preparation was carried out using SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA), and sequencing was performed using NextSeq500 (Illumina Inc., San Diego, CA, USA), generating 2×150 bp paired-end reads. Alignment of sequence reads, indexing of the reference genome (hg19), and variant calling were conducted using the GATK Best Practice pipeline (https://software.broadinstitute.org/gatk/best-practices/).
Identification of causative gene
Considering the AD pattern of inheritance, we filtered heterozygous variants (0.4<variant allele frequency [VAF]<0.6) that were shared by the five affected individuals and absent (VAF=0) in the three unaffected individuals as candidates. The variants were restricted to those with a population minor allele frequency <1% in the 1000 Genomes Project database [8], the Exome Aggregation Consortium database (ExAC; http://exac.broadinstitute.org/), or the Korean Reference Genome Database (KRGDB; http://152.99.75.168/KRGDB/). The variant identified in the segregation analysis was confirmed by Sanger sequencing using primers designed by the authors (available on request) in five individuals who underwent WES (II:4, II:6, III:16, III:20, III:21) as well as four additional family members with NSHL (II:7, II:11, II:13, III:22).
Functional studies
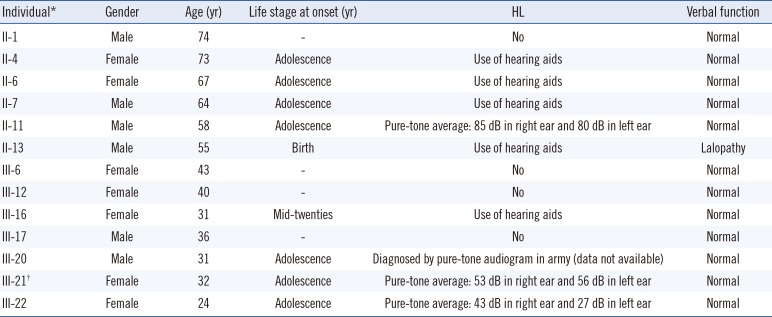
Following identification, the candidate variant of USH1C was introduced into a harmonin cDNA fragment encoding the full-length harmonin- a isoform and PDZ2 domain (amino acids 197 to 208; GenBank accession No. AF228924) and cloned into the pGST//2 vector. Recombinant proteins were expressed purified as described previously [9]. The differences in the measured affinity between the wild-type PDZ2 and the PDZ2 variant with cdh23 could be attributed to the inaccessibility of the binding site in the variant PDZ2 and this could result either from the presence of a bulky and polar cysteine residue instead of glycine at position 223 or from a conformational change of the PDZ2 domain induced by the variant. We therefore used circular dichroism spectra to compare the global secondary structure of the wild-type (WT) and variant PDZ2 domains. Circular dichroism experiments on the PDZ2 domains were performed using an Aviv 215 spectropolarimeter (Aviv Biomedical, Inc., Toms River, NJ, USA). Far-UV (195–260 nm) spectra were recorded at 20℃ in 50 mM Tris [pH 8], 200 mM KCl, and 0.5 mM Tris (2-carboxyethyl) phosphine (TCEP). The wavelength range used was 195–230 nm, with a band width of 5.0 nm.
To investigate the potential dominant negative effect of the harmonin variant, we examined two mutually nonexclusive possibilities. One is sequestration of partners due to the variant, and the other is the formation of hetero-oligomers between the WT and variant protein. Both the WT and variant full-length harmonin used in this study were individually purified and subsequently subject to size-exclusion chromatography (SEC) [9] to compare the elution volume of the analytes in solution (50 mM Tris [pH 8], 200 mM NaCl, and 0.1 mM TCEP) on the basis of their respective sizes.
Biolayer interferometry (BLI) experiments were performed using an Octet Red 384 instrument (Pall ForteBio, Fremont, CA, USA) [10]. Since the Octet instrument allows performing binding assays at 4℃ above the ambient temperature, the room temperature was maintained around 20℃ to perform the binding studies at 25℃. Streptavidin biosensors (Pall ForteBio) were loaded at room temperature with a biotin-tagged synthetic peptide comprising 12 amino acid residues, including the C-terminal PDZ-binding motif (PBM) sequence of cdh23. Reference sensors were loaded with biocytin. Both series of biosensors were then incubated in parallel for 2 min at 25℃ with proteins at different concentrations ranging from 2.2 to 60 µM in 50 mM Tris (pH 8), 200 mM KCl, and 0.5 mM TCEP. Signals measured on the reference sensors were subtracted from those measured on the peptide sensors. The affinities of the WT and variant PDZ2 were determined by analyzing the concentration-dependence of steady-state BLI signals.
RESULTS
Identification of USH1C variant
The process of segregation analysis using WES data is presented in Table 2. Two variants were filtered. One was a novel heterozygous missense variant (NM_005709.3:c.667G>T;p.Gly223Cys) of USH1C, which is a known gene related to HL. Sanger sequencing confirmed co-segregation of the USH1C variant with the NSHL phenotype in this family. The variant was identified in all affected participating family members, i.e., the five affected individuals (II:4, II:6, III:16, III:20, III:21) and four affected relatives (II:7, II:11, II:13, III:22); however, it was not present in the three unaffected family members (III:6, III:12, III:17) and in an additional unaffected individual analyzed by Sanger sequencing (II:1; Fig. 1F). All other variants of USH1C and genes known to be associated with HL were either classified as benign or likely benign or did not segregate with the NSHL phenotype in this family. The other variant was an ABCC8 synonymous variant (NM_000352.3:c.3918C>G;p.Leu1306=), which is listed in the dbSNP (rs372186045; https://www.ncbi.nlm.nih.gov/snp/rs372186045/), with a frequency of 0.26% in the East Asian population. Considering that the distance between the USH1C and ABCC8 variants is only 129 kb on chromosome 11, the synonymous ABCC8 variant may be in linkage disequilibrium with the USH1C variant.
Functional effect of the USH1C variant
The affinity of the WT PDZ2 for a biotinylated synthetic peptide containing the PBM of cdh23 (biotin-TtdsVIMESPLEITEL) was approximately 16-fold higher (Kd=37.6±4.1 µM) than that of the variant PDZ2 carrying the missense USH1C variant (Kd=621±88 µM; Fig. 2A). The different shapes of the curves obtained in the circular dichroism experiments indicated that the variant causes a conformational change in the PDZ2 domain (Fig. 2B).
In SEC experiments, WT full-length harmonin showed a single elution peak in the chromatogram, corresponding to the monomeric state of the protein, whereas the variant full-length harmonin showed a slightly leftward shift of the elution peak (Fig. 2C). When both WT and variant full-length harmonins were mixed and subjected to SEC, a more pronounced leftward shift of the peak was observed, accompanied by the appearance of another peak further leftward, indicating the elution of a higher molecular weight species. The difference in the elution volume, reflected in the differentially positioned peaks, potentially indicates the formation of a hetero-dimer between the WT and variant harmonins.
DISCUSSION
We report a large Korean family with a heterozygous USH1C missense variant and provide evidence suggesting a role for this gene in AD-NSHL. Stereocilia in the inner ear hair cell are coupled to one another by a variety of links, among which tip links, composed of cdh23 and cdh15, are the most crucial for mechano-electrical transduction [11]. Harmonin is found at the upper attachment of the tip link and forms a stable anchorage structure complex with cdh23 at the tip link of stereocilia via multivalent interactions [12]. Harmonin in retinal photoreceptors is localized at synaptic terminals and may play essential roles in the structural and functional unity of this synaptic junction; however, its function remains unknown [1314].
The PDZ domains in harmonin mediate the interaction between cdh23 and harmonin. These domains comprise 80–90 amino acid residues that contain six β-strands and two α-helices and have a single binding site in a groove between the αB and βB structural elements, with a highly conserved carboxylate-binding loop (R/K-XXX-G-Φ-G-Φ motif, where X is any amino acid residue and Φ is a hydrophobic residue) located before the βB strand (Fig. 2D) [15]. The p.Gly223Cys variant of harmonin affects the third residue of the GLGC motif in the carboxylate-binding loop of the PDZ2 domain, and it is predicted that this amino acid substitution disrupts the interaction between the PBM of cdh23 and the PDZ2 domain of harmonin [16]. In addition, the third glycine residue in the loop is fully conserved and adopts a left-handed α-helical conformation in all structures reported to date, and this residue may be important for determining the PDZ fold [17]. Therefore, the p.Gly223Cys variant in harmonin is likely to exert significant functional consequences by reducing the accessibility of the PDZ binding site to the target and inducing a structural modification.
The USH1C c.667G>T (p.Gly223Cys) variant was shown to be absent in 1,722 individuals in the KRGDB and 60,706 individuals in the ExAC. When this variant was classified according to the Sequence Interpretation Guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [7], it was interpreted as a likely PV (LPV) based on the following evidence: absent in population database (PM2), multiple lines of computational evidence supporting a deleterious effect on the gene (PP3), and co-segregation with disease in multiple affected family members (from supporting evidence PP1 to strong evidence with a large extent of segregation). If a well-established functional study showing a deleterious effect (PS3) could be conducted based on the present data, the variant classification might change from LPV to PV.
This study shows that a single PV in USH1C can cause disease in NSHL patients. The USH1C PVs reported thus far are mostly null variants (including nonsense, frameshift, and splicing mutations) on both gene copies with some exceptions: a compound variant heterozygous for a null and a missense mutation, a homozygous missense change variant, and a homozygous “leaky” splice-site mutation variant. AD transmission related with HL has yet to be described to the best of our knowledge [5]. Therefore, conventional genetic diagnostic approaches based on the well-known inheritance pattern of each gene in NSHL may fail to identify the real causative variant in a minor portion of NSHL patients.
 XML Download
XML Download