

 PDF
PDF ePub
ePub Citation
Citation Print
Print


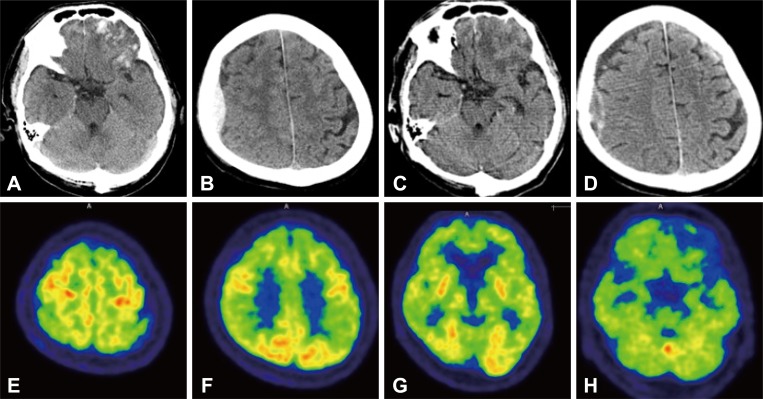
A 71-year-old male visited the emergency room with altered mentality. His co-workers found him lying unconscious at home on the day of the visit to the hospital. The patient was a general surgeon of our hospital who, just before the illness, performed his profession normally. At the time of arrival, neurological examination showed mental status as drowsy and Glasgow Coma Scale of 11 (E3V3M5). Brain CT revealed a skull fracture in the right occipital area, coup epidural hemorrhage in the right frontoparietal area, contrecoup contusions in the frontal and left anterior temporal area, and subarachnoid hemorrhage in the both frontal convexity (a traumatic coup-contrecoup pattern) (Fig. 1A and B).
The Korean version of Mini-Mental Status Examination (K-MMSE) score on the 15th day of hospitalization was 4 out of 30. A follow up brain CT showed resolving process of the hemorrhage (Fig. 1C and D). The patient did not remember the situation at the time of the incident, but traumatic brain injury (TBI) was suspected, considering the CT findings of the skull fracture and the coup-contrecoup pattern. Other investigations also did not reveal any abnormalities to explain the cause of the mental deterioration.
At 17 days after admission, we performed an 18F fluorodeoxyglucose positron emission tomography (FDG-PET), which showed decreased metabolism in both frontal, temporal, and parietal lobes symmetrically. The metabolism of the primary sensorimotor cortex, primary visual cortex, and subcortical structures were preserved, consistent with typical Alzheimer's disease (AD) (Fig. 1E-H). Two months after the trauma, the patient scored 13 points on a repeated K-MMSE. However, the follow up FDG-PET scan showed no improvement.
There are no consistent FDG-PET findings for TBI, due to the various mechanisms and imaging methods.1 In our case, PET findings were consistent with typical AD. Our patient showed no clinical signs of the disease until the accident. We presume that TBI triggered the existing subclinical AD.
The association between TBI and AD has been of interest since the observation of beta amyloid in 30% of brains who died after acute TBI.23 In addition to forming beta amyloid in the acute phase, TBI may also initiate neurodegeneration.4 Many epidemiological studies have confirmed the association between TBI and AD.5 The amyloid precursor protein is thought to accumulate in the axon damaged by TBI, which is abnormally cleaved to form beta amyloid. Tau phosphorylation, neurite degeneration, synapse loss and microgliosis are also observed, which may be related to neuroinflammation caused by TBI.678 Studies relating to the association between TBI and AD are ongoing, and the relationship is expected to become clearer in the future.
 XML Download
XML Download