

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Adrenocortical carcinoma (ACC) is an uncommon and heterogeneous endocrine malignant neoplasm with an estimated incidence of 0.5–2.0 cases per million per year (12). It usually occurs in adults with a peak incidence in the fifth decade of life; females are more commonly affected than males (3). Functional ACC occur at a rate of approximately 42%–57% of all ACCs and more frequently arise in women (4). In addition to conventional ACC, distinct histological subtypes, such as oncocytic, myxoid, and sarcomatoid variants, have been described (5).
The myxoid variant of ACC was first reported in 1979 by Tang et al. (6). A total of 42 cases of myxoid ACC have been reported to date (67891011121314151617). The sarcomatoid variant of ACC was first reported in 1987 by Okazumi et al. (18). Only 13 cases of sarcomatoid ACC, including adrenocortical carcinosarcoma, have been reported to date (18192021222324252627282930).
Patients with ACC generally show an unfavorable prognosis and a marked inter-individual variability in disease progression, recurrence, and overall survival (12). Because of the difficulty in differentiating the benign from malignant adrenocortical tumors, various multi-parametric diagnostic algorithms, such as the Weiss, Hough, van Slooten, modified Weiss scoring systems, and reticulin algorithm, have been used (3132). Although the Weiss score is considered to be a simple and reliable system for predicting malignant cases amongst conventional ACC, it has some limitations in cases of oncocytic, myxoid or sarcomatoid variant ACC, and also in pediatric adrenocortical neoplasm (34).
The rarity of the myxoid or sarcomatoid variants of ACC precludes a valid prognostic assessment in these patients. In addition to tumor stage and distant metastasis, the Ki-67 labeling index has been reported as a reliable tool for prognostic assessment in patients with ACC (12).
The aim of the present study was to describe the clinicopathological characteristics of 9 cases of myxoid or sarcomatoid variants of ACC treated at a single tertiary care center using the Weiss scoring system, Ki-67 labeling index, and reticulin framework alteration algorithm. We compared the prognosis for these rare variants of ACC with that of conventional ACC and identified variables associated with poor clinical outcomes.
MATERIALS AND METHODS
Study subjects
This retrospective study included 44 cases of ACC confirmed on immunohistochemical (IHC) examination at the Asan Medical Center, Seoul, Korea between January 1997 to May 2014, and for whom the primary adrenal tumor specimens were available for pathological review. There were 18 cases of conventional ACC, 17 cases of the oncocytic variant, 8 cases of the myxoid variant, and 1 case of the sarcomatoid variant of ACC. Patients younger than 18 years or those having the oncocytic variant ACC which uses other diagnostic algorithm besides Weiss scoring system were excluded from this study.
We categorized the patients into 2 groups according to the ACC subtype, i.e., 14 cases of conventional ACC and 9 cases of variant ACC (including myxoid and sarcomatoid). Various clinicopathological variables, such as age at diagnosis, sex, initial presentation (i.e., incidentaloma), size and weight of the primary tumor, status of hormone secretion, tumor stage, Weiss score, Ki-67 labeling index, presence of venous tumor thrombus, mode of metastasis, and therapeutic modality, were assessed.
The European Network for the Study of Adrenal Tumors Classification (ENSAT 2008) was used for tumor staging (33). Overall survival from the date of the initial surgery was determined.
Histopathological evaluation
All hematoxylin-and-eosin-stained slides of primary adrenal tumor specimens were reviewed by an experienced endocrine pathologist to assess malignancy using the Weiss scoring system (34). Tumors with 10% or more myxoid or sarcomatoid areas were included as rare variants of ACC in this study. Representative paraffin blocks of myxoid ACC were available for histochemical staining (Alcian-Blue pH 2.5, periodic acid-Schiff [PAS], and mucicarmine). Reticulin fiber alteration was evaluated for the 8 myxoid variants using a commercially available silver impregnation-based kit (Roche reticulum II staining kit; Ventana Medical Systems, Tucson, AZ, USA). Quantitative and qualitative assessment of the “altered” reticulin pattern was performed and compared with the “intact” fishing net-like reticulin architecture of the normal adrenal gland (32).
The Ki-67 IHC staining was performed for all 44 ACC cases using a Benchmark device (Ventana Medical Systems). Whole-tissue sections of 4-μm thickness obtained from formalin-fixed, paraffin-embedded specimens were transferred to poly-L-lysine-coated adhesive slides and dried at 74°C for 30 minutes. After heat epitope retrieval for 1 hour in ethylene diamine tetraacetic acid, pH 8.0, slides were incubated with Ki-67 antibody (clone MIB-1, 1:200 dilution; DAKO, Glostrup, Denmark). The slides were subsequently incubated with a reagent from the UltraView Universal DAB kit (Ventana Medical Systems) and counterstained with Harris hematoxylin. Negative controls were prepared by omitting the primary antibody, and positive controls were prepared using tonsil tissue. The Ki-67 labeling index was evaluated semi-quantitatively by selecting the hottest spot with positively-stained tumor cells. In all cases, 5–10 high-power fields were selected; a minimum of 1,000 cells were independently evaluated. The number of Ki-67 positive cells per 100 tumor cells was designated as the Ki-67 labeling index.
Statistical analysis
All statistical analyses were performed using R (version 3.1.0) and the R library package (R Foundation for Statistical Computing, Vienna, Austria; http://www.R-project.org). Continuous variables are presented as medians (interquartile range [IQR]). Categorical variables are presented as frequencies (percentages). The Student's t-test and Wilcoxon rank-sum test were used to assess between-group differences with respect to continuous variables. The χ2 test or Fisher's exact test were used to compare categorical variables. Kaplan-Meier survival curves were constructed; between-group differences in survival were assessed by log-rank test. Hazard ratios (HR) and 95% confidence intervals (CI) for death or recurrence were calculated using Cox proportional hazard model. A P value of < 0.05 was considered statistically significant. All P values were 2-sided.
RESULTS
Clinicopathological parameters of myxoid or sarcomatoid variant ACC
Nine patients (20%, 9/44) in our study series had rare histological subtypes of ACC (8 myxoid variants and 1 sarcomatoid variant). Clinicopathological characteristics of these cases are summarized in Table 1. The mean age was 45 years; 4 patients were female. Mean primary tumor size was 12.9 ± 5.7 cm; mean primary tumor weight was 702.4 ± 785.9 g. Of the 3 patients for whom functional data was available, 2 showed Cushing's syndrome. Of the 9 patients, 7 presented with advanced tumor stage (stage III/IV). Eight patients eventually developed distant metastasis. The liver was the most common site of distant metastasis followed by the lungs. The mean Weiss score was 5.0 ± 1.7 points and Ki-67 labeling index was 15.6% ± 16.4%. The Ki-67 labeling index ranged from 1% to 48% in the hottest spot. Four patients showed a venous tumor thrombus. These clinicopathological parameters were similar to those of conventional ACC (Table 2).
Table 1
Clinicopathological parameters of patients with myxoid or sarcomatoid variants of ACC

Table 2
Clinicopathological parameters of ACCs disaggregated by histological subtype
The histological characteristics according to the Weiss scoring system and the modified Weiss scoring system for myxoid or sarcomatoid variants of ACC are summarized in Table 3. The most common histological feature of the Weiss scoring system was the presence of necrosis in 8 cases (88.9%), followed by sinusoidal invasion in 7 cases (77.8%), and capsular invasion in 7 cases (77.8%). The least common histological features of the Weiss scoring system were the presence of atypical mitotic figures and diffuse architecture in 2 cases each (22.2%). The mean Weiss score was 5.0 ± 1.7 and the mean modified Weiss score was 3.89 ± 1.60. According to the modified Weiss scoring system, the features of myxoid ACC case 1 were not consistent with a malignant status (Table 3).
Table 3
Histological characteristics according to the Weiss scoring system of myxoid or sarcomatoid variant ACC

Histological features of myxoid or sarcomatoid variant ACC
Grossly, the myxoid or sarcomatoid variant ACCs examined in our current study were variably encapsulated masses with lobulation, focally gelatinous and with translucent cut surfaces (myxoid variant, Fig. 1A), or yellow-tanned and partially white fleshy cut surface (sarcomatoid variant, Fig. 1E). These tumors ranged in size from 3.8 to 22.0 cm in their greatest dimension and weighed between 38.5 and 2,292.0 g. These showed immunopositivity for inhibin, synaptophysin, melan-A, and immunonegativity for chromogranin at the time of initial diagnosis, which is supportive of the diagnosis of ACC. Microscopically, the extent of myxoid or sarcomatoid change ranged from approximately 10%–75% in the 8 cases of myxoid ACC and 60% in the lone case of sarcomatoid ACC. The myxoid areas showed various growth patterns, including trabecular, inter-anastomosing cords, small clusters and microcystic patterns in a loose myxoid stromal component background (Fig. 1B). Tumor cells in the myxoid areas were relatively smaller than those in the non-myxoid areas and showed mild nuclear atypia, hyperchromatic nuclei, inconspicuous nucleoli, and scant eosinophilic cytoplasm.
Fig. 1
Histological features of myxoid (A-D) and sarcomatoid (E, F) variants of ACC. Grossly, the myxoid variant ACC shows a variegated cut surface with yellowish tan, necrotic, focally gelatinous and hemorrhagic foci (A). Tumor cells of myxoid ACC show various growth patterns, including inter-anastomosing cords, small clusters, and microcystic patterns (B) in an Alcian-Blue positive myxoid stroma background (C) and also show reticulin network alterations (D). Grossly, the sarcomatoid variant of ACC shows a variegated cut surface with yellowish tan, necrotic, and partially white fleshy foci (E). Tumor cells of the sarcomatoid variant ACC show a diffuse growth pattern with mainly spindle cells (F).
ACC = adrenocortical carcinoma.

One case of myxoid ACC (case 7, Table 1) showed adipose metaplasia, which mimicked immature adipocytes or lipoblasts. The extracellular myxoid stromal component showed positive staining for Alcian-Blue (Fig. 1C) and negative staining for PAS and mucicarmine, which suggests the possibility of an acidic mucopolysaccharide substance. There was no evidence of any intracellular myxoid substance. A dense myxoid stromal component occasionally mimicked chondroid stromal changes. All myxoid variants showed a disrupted reticulin network, which contrasted with the intact reticulin fibers in the adjacent normal adrenal parenchyma. Areas of altered reticulin framework showed both an extensive loss of reticulin fibers (Fig. 1D) and irregularly thickened or frayed reticulin fibers in all cases with a heterogeneous distribution. Sarcomatoid ACC showed a diffuse growth pattern and consisted of mainly spindle cells (Fig. 1F) with occasional epithelioid cells and giant cells. There was no evidence of a heterologous sarcomatous component akin to that seen in rhabdomyosarcoma, osteosarcoma, or chondrosarcoma.
Clinicopathological parameters associated with overall survival
On univariate analyses, the tumor subtype (P = 0.070) and the Ki-67 labeling index (P = 0.060) showed marginally significant association with overall survival (Table 4). Log-rank test revealed a similar marginal association of overall survival with the tumor subtype (P = 0.060) and the Ki-67 labeling index (P = 0.050) (Fig. 2A and B). Median survival for conventional ACC was 1.9 years, as opposed to 0.7 years for patients with myxoid or sarcomatoid ACC. Median survival in patients with a Ki-67 labeling index < 10% was 3.2 years, as compared to 0.7 years for patients with a Ki-67 labeling index > 10%. After adjusting for age and sex, rare variants of ACC (HR, 3.59; 95% CI, 1.13–11.38; P = 0.030), and the Ki-67 labeling index (HR, 3.97; 95% CI, 1.18–13.41; P = 0.030) were found to be independent predictors of overall survival in ACC patients on multivariate analysis (Table 4).
Table 4
Clinicopathological parameters associated with overall survival

Fig. 2
Overall survival of patients with adrenocortical carcinoma disaggregated by tumor subtype (A) and Ki-67 labeling index (B) by the log-rank test. Presence of myxoid or sarcomatoid histological features (A) and an increased Ki-67 labeling index (B) show a similar marginally significant association with overall survival on the log-rank test.
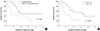
DISCUSSION
In the present study, we first assessed the clinical impact of the rare myxoid or sarcomatoid features in ACC by comparing the prognosis of these rare variants with that of the conventional ACC. Although our analysis was limited by its retrospective design, the small number of patients analyzed, and the potential referral bias of a single tertiary center, we included only validated rare variants of ACC, i.e., those with ≥ 10% myxoid or sarcomatoid component, for the purpose of this analysis. The prognosis of the myxoid or sarcomatoid variant ACC has remained uncertain because of the small series of reported cases, limited follow-up periods and insufficient clinical data. For cases of myxoid ACC, similarly aggressive (1112) or more aggressive clinical behavior than conventional ACC has been previously reported (16).
In cases of sarcomatoid ACC, a very poor prognosis with frequent metastases was reported, with the majority of patients dying within 3–12 months after surgical treatment (2529). The longest reported postoperative survival with no evidence of metastasis was 17 months in a 58-year-old male with adrenal sarcomatoid carcinoma described by Mark et al. (29).
The mean Ki-67 labeling index for myxoid or sarcomatoid ACC (15.6%) was higher than that for the conventional ACC cases (8.21%) in our series, but lower than those reported previously (31.8%) for adrenocortical tumors with myxoid features by Papotti et al. (13). Patients in our present study with a rare variant ACC or an increased Ki-67 labeling index (> 10%) showed a similar median survival of 0.7 and 0.8 years, respectively. After adjusting for age and sex, the presence of rare variant ACC (HR, 3.59; 95% CI, 1.13–11.38; P = 0.030) and an increased Ki-67 labeling index (HR, 3.97; 95% CI, 1.18–13.41; P = 0.030) was an independent predictor of overall survival in patients with ACC (Table 4). Thus, histological recognition and sub-classification of myxoid or sarcomatoid features, and Ki-67 IHC staining might be useful for predicting the prognosis in ACC patients. Myxoid or sarcomatoid histological features in ACC ranged widely from 10% to 75% in our present analyses, which suggests the need for a thorough histopathological examination for large bulky tumors to arrive at a correct diagnosis.
The Weiss scoring system has been the most popular scoring system of ACC because of its reproducibility and simplicity (34), but it has some limitations for use in pediatric, oncocytic and rare myxoid variants of ACC. Tumor cells of myxoid ACC demonstrate relatively mild nuclear atypia, a lack of diffuse growth pattern and equivocal sinusoidal invasion compared to conventional ACC. The most common histological feature of the Weiss scoring system in our present study series was the presence of necrosis (88.9%). More than half (75%) of the variant ACC cases with predominant myxoid features showed a Weiss score of 3 or 4. Moreover, 1 case of myxoid ACC in our series was less of histological features of malignancy according to the modified Weiss scoring system, suggesting a risk of under-diagnosis of ACC in the case of adrenocortical tumors with myxoid features. However, all myxoid ACC analyzed herein demonstrated an altered reticulin framework associated with at least 1 out of 3 of the following parameters: necrosis, high mitotic rate, or vascular invasion, thus satisfying the “reticulin algorithm” for defining malignancy in rare myxoid variants of ACC (32). Reticulin fiber architecture was found to be altered both quantitatively and qualitatively with a heterogeneous distribution in our current study cases. This finding is consistent with those reported elsewhere (13). Extensive loss of reticulin fibers was more frequently observed than irregularly thickened and frayed reticulin fibers in this study.
Here, we present the fifth reported case of myxoid ACC with adipose metaplasia to date. Only 4 cases of myxoid ACC with adipose metaplasia are on record (91617) including the first case report by Izumi et al. (9). Adipose metaplasia in myxoid ACC may represent a reactive degenerative or metaplastic process in the tumor cells. Izumi et al. (9) described various stages of lipid accumulation in tumor cells mimicking mature adipocytes with a vacuolated cytoplasm in the case of myxoid ACC with extensive adipose metaplasia, suggesting a possibility of metaplastic process. In our current cases, foci of mature adipocytes of variable sizes were scattered throughout the tumor in a pattern similar to that reported previously (91617).
The differential diagnoses for rare myxoid or sarcomatoid ACC include metastatic tumors with myxoid or sarcomatoid features, and various primary retroperitoneal myxoid tumors, including chordoma, myxoma, extraskeletal myxoid chondrosarcoma, lipoma, liposarcoma, benign or malignant nerve sheath tumors, myxoid leiomyoma, myxoid leiomyosarcoma, gastrointestinal stromal tumor, and primary retroperitoneal sarcomas. A comprehensive histopathological examination of such tumors, a full assessment of the previous medical history, clinicoradiological correlation, and IHC staining panel can be useful in distinguishing between these various lesions.
In conclusion, we validated the utility of a reticulin algorithm for predicting malignancy of myxoid variant ACC. The presence of rare myxoid or sarcomatoid histological features, or an increased Ki-67 labeling index may be associated with a more aggressive clinical behavior and poor overall survival in ACC patients; however, a more extended further study is required.
 XML Download
XML Download