

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Inflammatory bowel disease (IBD) is chronic inflammation of the gastrointestinal tract, characterized by unpredictable exacerbation and remission due to multiple factors, such as genetic factors, the intestinal epithelial barrier, and abnormal immune response.1 IBD is broadly divided into ulcerative colitis, Crohn's disease, and indeterminate colitis, but each disease has a heterogeneous course with differences in location, behavior, and inflammation severity. Even patients in the same category have different prognoses. The incidence of IBD has increased considerably in the last decade. The peak age of onset is adolescence. Many anti-inflammatory drugs including steroids, 5-aminosalicylic acid, antibiotics, immunosuppressive agents, or tumor necrosis factor-α (TNF-α) inhibitors have been used for treating IBD. However, these current drug therapies can temporarily relieve acute inflammation, but cannot alter prolonged inflammation or aggravated progression. Therefore, a need exists for more effective drugs for treating IBD.2
Insulin-like growth factor-binding protein-3 (IGFBP-3) is a component of the insulin-like growth factor (IGF) system linked to the control of cellular growth and differentiation. Seventy-five to ninety percent of circulating insulin-like growth factor-1 (IGF-1) is bound to IGFBP-3.3 The anti-inflammatory effects of IGFBP-3 are mediated by inhibition of nuclear factor-κB (NF-κB) signaling through an IGF-independent mechanism. IGFBP-3 reduces TNF-α activity and induces apoptosis in carcinoma cells.4 Exogenous IGFBP-3 or upregulated IGFBP-3 reduces synovial joint inflammation and bone destruction in murine models of rheumatoid arthritis.5 Exogenous human IGFBP-3 inhibits airway inflammation; thus, opening up the possibility of using it as a therapeutic agent for bronchial asthma.6 These abilities of IGFBP-3 suggest that it can be used for inhibiting inflammation in patients with IBD.
Many inflammatory cytokines are associated with IBD pathogenesis, although the complete mechanisms associated with these cytokines have not been fully elucidated yet.7 Hence, the regulation of some widely studied cytokines, like TNF-α and cyclooxygenase (COX)-2, have been suggested as a treatment protocol in IBD.89 TNF-α inhibitor has received much attention because of their ability to induce and maintain remission.8 The regulation of anti-inflammatory cytokines is the key point in managing IBD. Studies have confirmed that patients with IBD have significantly reduced levels of serum IGF-1 and IGFBP-3.1011 Treatment with steroids or infliximab normalizes serum levels of IGFBP-3.1112 The underlying mechanism of IGFBP-3 is unknown, but we studied in vitro and in vivo effects of IGFBP-3 in a rat small intestinal epithelial cell line (IEC-6) and a colitis mice model, respectively. These results will contribute to a new therapeutic protocol for patients with intractable IBD.
METHODS
Intestinal epithelial cell culture and LPS-treated stress
IEC-6 is a rat ileal epithelial cell line obtained from the Korean Cell Line Bank (No. 21592). Lipopolysaccharides (LPS, L4524) from Escherichia coli 055:B5 were purchased from Sigma (St. Louis, MO, USA). Ad/LacZ (adenoviral vectors expressing E. coli β-galactosidase) and adenoviral vector system expressing IGFBP-3 (Ad/IGFBP-3) were made using the AdEasy vector system (Quantum Biotechnologies, Montreal, CA, USA).13 To create Ad/LacZ, a SalI-NotI fragment from pcDNA3.1/LacZ was ligated into the cytomegalovirus (CMV) vector. As for Ad/IGFBP-3, NotI-XbaI fragments from pcDNA3/IGFBP-3 DNA were ligated into CMV.14 These recombinations and the pAdEasy-1 viral backbone were created by homologous recombination in E. coli (strain BJ5183). Recombinant DNA of adenovirus was verified and transferred to DH5α (E. coli strain). After linearizing by PacI-digested DNA, the recombinant CMV-cDNA was transferred to QBI293A (human embryonic kidney cells) using the calcium phosphate transfection method (Promega, Madison, WI, USA). Recombinant viruses were amplified in QBI293A and purified by CsCl centrifugation (100,000 g at 4°C for 20 hours). The adenoviruses were titrated by plaque analysis using serial dilution infection. The infection was measured by the multiplicity of infection (MOI) in subconfluent cells in % fetal bovine serum (FBS) medium.
The cells were cultured in the Dulbecco's Modified Eagle medium (DMEM; Life Technologies, Grand Island, NY, USA), supplemented with L-glutamine (300 μg/mL), penicillin (100 U/mL), streptomycin (100 μg/mL), and 5% FBS. One million cells were seeded in 10 mL culture dishes at 37°C and incubated overnight in a humidified atmosphere of 5% carbon dioxide. Once they reached confluence, the cells were detached from the plate by trypsinization, washed, and replated in DMEM. In this study, we used cells between passages 5 and 10. IEC-6 cells were divided into 6 groups: 3 normal groups without LPS stress and 3 with LPS stress. To establish the LPS stress model, when the cells reached over 90% confluence, the medium was replaced with DMEM containing 0.5% FBS and the cells were incubated for 18 hours. After a starvation period, the cells were infected with 100 MOI of Ad/LacZ or Ad/IGFBP-3. After a 2 hours incubation, the medium was replaced with DMEM containing 0.5% FBS, and the cells were stimulated with 10 μg/mL LPS for 24 hours.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability was measured using the MTT assay as described previously. The MTT solution (Sigma) was used to evaluate IEC-6 cell viability or cytotoxicity after treatments. The treated cells were distributed in 96-well plates. After removing the DMEM, 20 μL of MTT solution (200 μg/mL) was added to each well and incubated for 3 hours at 37°C. The supernatant was then removed and 150 μL of dimethyl sulfoxide (DMSO) was added. After vigorous shaking to dissolve the formazan crystals, absorbance of the wells was measured using a spectrophotometer (ELX 800; Bio-Tek Instruments, Winooski, VT, USA) at a wavelength of 540 nm. Cell viability was calculated using the following equation:
Measurement of reactive oxygen species (ROS) formation
At the indicated time, ROS generation was analyzed by FACScan flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA) using a 2′,7′-dichlorofluorescin diacetate (DCFH-DA) (Fluka, Molecular Probes, Eugene, OR, USA) fluorescent probe. IEC-6 cells in 10 mL plates were incubated with 5 μmol/L DCFH-DA for 30 minutes at 37°C in the dark. After washing two times with phosphate buffered saline (PBS), the cells were collected by centrifugation (900 rpm for 4 minutes). The mean fluorescence at the emission wavelength of 520 nm was measured after excitation at 480 nm. The data were analyzed by Becton Dickinson Partec software and expressed as a representative histogram.
Western blot assay
Antibodies against COX-2 (sc-1745), interleukin (IL)-1β (sc-7884), TNF-α (sc-1350), NF-κB (sc-109), IGFBP-3 (sc-9028), and lamin B (sc-6216) were purchased from Santa Cruz (Santa Cruz, CA, USA). Antibodies against β-actin (#A-2066) were purchased from Sigma, phospho-p38 MAPK (#9211) and phospho-ERK (#9101) from Cell Signaling (Beverly, MA, USA), and Anti-Cu/Zn (#SOD-100) and Anti-Mn SOD (#SOD-110) were from Stressgen (Ann Arbor, MI, USA).
Western blotting was performed according to the standard protocol.13 Proteins were extracted from cells by immersing the cells in non-denaturing lysis buffer (Tris-HCl, NaH2PO4, NaCl, Triton X-100, sodium pyrophosphate, pH 7.5) for 10 minutes on ice. Cellular debris was eliminated by centrifugation. The samples were stored at −80°C. The protein concentrations were quantified using the Bradford dye-binding assay kit (Bio-Rad, Hercules, CA, USA). Twenty micrograms of protein were diluted, loaded, and separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) without reducing conditions. Proteins were electroblotted onto Hybond filters (Amersham Biotech, Piscataway, NJ, USA). Membranes were blocked with 5% nonfat milk/Tris-buffered saline and Tween (TBST) for 30 minutes at room temperature and incubated with the appropriate antibody for 2 hours. Protein bands were detected using enhanced chemiluminescence (NEN, Boston, MA, USA).
Animals
Eight-week old female mice (C57BL6) weighing 25–30 g were purchased from Samtako (Osan, Korea). All mice were placed in plastic cages and had access to water and food, under 12-hour dark/light cycles at controlled temperatures (25°C–27°C).
Induction of colitis and treatment
Dextran sodium sulfate (DSS) was purchased from MP Biomedical (MW: 36,000–50,000; Solon, OH, USA). The mice were sorted into 4 experimental groups: 1 control group and 3 colitis groups. To induce colitis in mice,15 the mice were given drinking water mixed with 3% DSS for 7 days (day 1 to 7). On day 7, the DSS-mixed water was replaced with purified water. Meanwhile, the control group received normal water. The mice were monitored carefully and their body weights were measured daily. Ad/IGFBP-3 or Ad/LacZ was administered into the rectosigmoid colon by enema on day 3 and day 6. For this procedure, mice were anesthetized using diluted ketamine without any complication. After PBS enema and washing, 200 μL of Ad/LacZ or Ad/IGFBP-3 (1011 pfu) were added using a 5 Fr catheter (inserted up to 4 cm in length from anus). The other 2 groups (control and DSS colitis) were intracolonically injected with normal saline using the same protocol. On day 10, the mice were sacrificed by vertebral dislocation, the colons were then removed and rinsed in PBS. To evaluate colitis severity, colon length was measured macroscopically, and histological studies, including microscopic scoring, were performed. Histological evaluation was performed by two scoring systems. The crypt scoring was based on pathological changes as follows: score 0 = intact crypt, score 1 = loss of one-third of the crypts, score 2 = loss of two-thirds of the crypts, score 3 = loss of whole crypt with mild inflammatory cell infiltration, score 4 = erosions with marked inflammatory cell infiltration. Involvement scoring means percentage of total mucosal slide involved as follows: score 0 = normal colonic mucosa, score 1 = 1%–25% involvement, score 2 = 26%–50% involvement, score 3 = 51%–75% involvement, score 4 = 76%–100% involvement.16
Statistical analysis
Results were expressed as the means ± standard error, where applicable. Data analyses were performed using unpaired Student's t-test (parametric) and the Mann-Whitney U test (nonparametric) for comparison. P value < 0.05 was considered statistically significant. SPSS software v21.0 (IBM, Armonk, NY, USA) was used for statistical analysis.
RESULTS
Exogenous IGFBP-3 expression in the mouse colon by the intracolonic administration of adenoviral particles
To confirm the expression of Ad/LacZ or Ad/IGFBP-3 in mouse colonic mucosa, mice were administered with the adenoviral particles through three administration routes: tail vein injection, peritoneal injection, and enema. The same amount of Ad/LacZ (1011 pfu, PBS dilution) was injected into the tail vein (50 μL) and intraperitoneal space (100 μL), and infused into rectosigmoid colonic lumen (200 μL) with an enema using a 5 Fr catheter. The 4 groups of animals including the control group were sacrificed after 3 days. The liver and colon were isolated and stained to detect the level of LacZ expression (Fig. 1A). We found that the rectal route was the most effective method of administration for colon-specific expression. The same protocol was applied for Ad/IGFBP-3 administration. Immunohistochemical staining was performed using an IGFBP-3 antibody purchased from Bioworld Technology (Louis Park, MN, USA). Comparing the 4 groups for IGFBP-3 expression (Fig. 1B), the intracolonic administration was the most effective method for expressing IGFBP-3 using the adenoviral expression system in mice.
Fig. 1
The organ staining for LacZ expression and the tissue immunohistochemistry staining for IGFBP-3 expression. (A) Rectal administration is the most effective method to express of LacZ in the colon (up; liver, down; colon). (a) Control, (b) intraperitoneal administration, (c) intravenous administration, (d) rectal administration. (B) The immunohistochemistry stain for detecting IGFBP-3 expression shows rectal administration is the most effective method. (a) Control, (b) intraperitoneal administration, (c) intravenous administration, (d) rectal administration (× 200).
IGFBP-3 = insulin-like growth factor-binding protein-3.

Increased IGFBP-3 expression inhibits LPS-induced cell cytotoxicity in IEC-6 cells
IEC-6 cells were sorted into 6 groups. To determine the effect of adenovirus infection on IEC-6 cells without LPS injury, two cell groups were treated with Ad/LacZ and Ad/IGFBP-3 viral particles, respectively. Cell viability or cytotoxicity was measured using an MTT assay 24 hours after treatment. The adenoviral infection had no cytotoxic effect on the cells without LPS stimulation (i.e., control, Ad/LacZ, and Ad/IGFBP-3 treated groups) (Fig. 2A). On the contrary, decreased cell viability was observed in the other 3 LPS-treated groups. However, pretreatment with Ad/IGFBP-3 significantly reduced LPS-induced cytotoxicity (Fig. 2A, P = 0.001).
Fig. 2
MTT assay and ROS formation in IEC-6 cells. (A) Cell viability was measured using the MTT assay after 24 hours of LPS stress (10 μg/mL) in IEC-6. Cell viability = A treatment/A control. The values were reported as statistical mean. Control, 1.0000; Ad/LacZ, 0.9976; Ad/IGFBP-3, 0.9965; LPS, 0.9288; LPS + Ad/LacZ, 0.9344; LPS + Ad/IGFBP-3, 0.9484. LPS + Ad/IGFBP-3 group shows higher viability than other two injury groups (LPS and LPS + Ad/LacZ). The values were reported as the mean ± SE. The LPS + Ad/IGFBP-3 group was compared to LPS + Ad/LacZ group. P < 0.05 was calculated using unpaired Student's t-test. (B) Flow cytometric histogram shows the high levels of ROS formation in response to LPS stress. The Ad/IGFBP-3 therapy reduced the ROS formation by a left-shift. The representative data were presented from ten independent experiments were performed.
MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, ROS = reactive oxygen species, IEC-6 = intestinal epithelial cell 6, LPS = lipopolysaccharides, Ad/IGFBP-3 = adenoviral vector system expressing insulin-like growth factor-binding protein-3, SE = standard error.

IGFBP-3 expression lowers the level LPS-stimulated IEC-6
ROS levels were detected using the fluorescent probe DCFH-DA. After 24 hours of 10 μg/mL of LPS stress, LPS-induced ROS formation was observed through increased fluorescence levels compared to levels in the three non-treated groups. There was no significant difference among the control, Ad/LacZ, and Ad/IGFBP-3 groups without LPS treatment. Compared to the 2 LPS-treated groups, pretreatment with Ad/IGFBP-3 showed marked protective effects with reduced ROS production in cells (Fig. 2B).
IGFBP-3 suppresses the expression of inflammatory cytokines
Cells were exposed to 10 μg/mL LPS for 24–48 hours. Inflammatory cytokines such as COX-2 or IL-1 were expressed by LPS stimulation in IEC-6 cells (Fig. 3A). Superoxide dismutase (SOD) is an important antioxidant enzyme that catalyzes and removes superoxide radicals generated by an inflammatory response. The expression of SOD enzyme and protein in the LPS-treated cells was gradually downregulated as time elapsed. In contrast, pretreating IEC-6 cells with Ad/IGFBP-3 sustained the level of Cu/Zn and Mn SOD (Fig. 3B). We next examined whether LPS-stimulated NF-κB would be influenced by IGFBP-3. Ad/IGFBP-3-treated cells showed reduced phospho-p38 MAPK, phospho-ERK (Fig. 3C), and nuclear NF-κB (Fig. 3D) levels in response to LPS. Upon LPS stimulation, the expression of pro-inflammatory cytokines IL-1β, COX-2, and TNF-α was upregulated. However, treatment with Ad/IGFBP-3 inhibited the expression of these cytokines (Fig. 3E). Phospho-IκB-α levels were also reduced by Ad/IGFBP-3 treatment (Fig. 3F). These results suggest that IGFBP-3 expression by Ad/IGFBP-3 suppresses inflammatory responses in LPS-treated IEC-6 cells.
Fig. 3
The Western blots in IEC-6 cells. (A) COX-2 and IL-1β levels in total cell lysate was determined using Western blot analysis. The cells were treated with LPS (0–10 μg/mL) for 24 and 48 hours. The expression of both COX-2 and IL-1β are increased according to cell damage or exposure duration. (B) The protein levels of Cu/Zn SOD and Mn SOD as an antioxidant enzyme were examined. The expressions were checked at 24 and 48 hours after LPS stress (10 μg/mL). Pretreated Ad/IGFBP-3 group maintained the levels of SOD enzyme. (C) Phospho-ERK and Phospho-p38MAPK signaling were measured. The adenoviral vector Ad/IGFBP-3 reduced the expression of P-p38MAPK and P-ERK. (D) The NF-κB activity was measured. The adenoviral vector Ad/IGFBP-3 reduced translocation of NF-κB from the cytosol to the nucleus. (E) Total cell lysate was measured using Western blot analysis. Pretreatment with Ad/IGFBP-3 reduced the expression of these pro-inflammatory cytokines (COX-2, IL-1β, TNF-α). (F) Treated with Ad/IGFBP-3 reduced the expression of phospho-IκB-α.
IEC-6 = intestinal epithelial cell 6, SOD = superoxide dismutase, LPS = lipopolysaccharides, IL = interleukin, COX = cyclooxygenase, Ad/IGFBP-3 = adenoviral vector system expressing insulin-like growth factor-binding protein-3, NF-κB = nuclear factor-κB, TNF-α = tumor necrosis factor-α.

Ad/IGFBP-3 treatment ameliorates the severity of DSS-induced colitis
The inflammation severity can be characterized by weight loss and shortened colonic length in DSS-induced colitis mice.17 The range of measured body weight of mice was 25–30 g on day 1, and there were no significant median weight differences among the groups. Sixteen mice were divided into 4 groups, and the mean values were compared among each group. DSS-induced colitis groups were characterized by loss of body weight (Fig. 4). On day 10, statistically significant differences in body weight measurements between the control and colitis groups were obtained, but the Ad/IGFBP-3 treated group showed a significant improvement in weight gain compared to the Ad/LacZ-treated group (Fig. 4, P = 0.028). After the end of the experiment (day 10), all mice were sacrificed by cervical dislocation. The colonic lengths were measured and the longest and the shortest lengths from each group were excluded. In the DSS-induced colitis groups, significant shortening of the colonic length was observed. This shortening was suppressed by IGFBP-3 expression through Ad/IGFBP-3 treatment (Fig. 5, P = 0.032).
Fig. 4
The change of body weights. On day 3 and 6, 1 × 1011 pfu (200 μL) Ad/LacZ or Ad/IGFBP-3 was administered per rectal. Body weights were examined on the same time of the day. The DSS + Ad/IGFBP-3 group compared to the DSS + Ad/LacZ group. Pretreated Ad/IGFBP-3 group shows higher body weights than Ad/LacZ treated group's one on day 10 (P = 0.028). Mean ± SE. P < 0.05 was calculated by unpaired Student's t-test.
Ad/IGFBP-3 = adenoviral vector system expressing insulin-like growth factor-binding protein-3, DSS = dextran sodium sulfate, SE = standard error.

Fig. 5
The change of colonic length. Colons were harvested on day 10, and colonic lengths were measured from the cecum to the rectum. The Ad/IGFBP-3-treated group compared to the DSS group or Ad/LacZ-treated group. Pretreated Ad/IGFBP-3 group shows higher colonic length than the other two group's one significantly (aP = 0.016, bP = 0.032). Statistical mean. aP < 0.05 and bP < 0.05 were calculated by unpaired Student's t-test.
Ad/IGFBP-3 = adenoviral vector system expressing insulin-like growth factor-binding protein-3, DSS = dextran sodium sulfate.

Ad/IGFBP-3 treatment provides a protective effect in the colon of DSS-treated mice
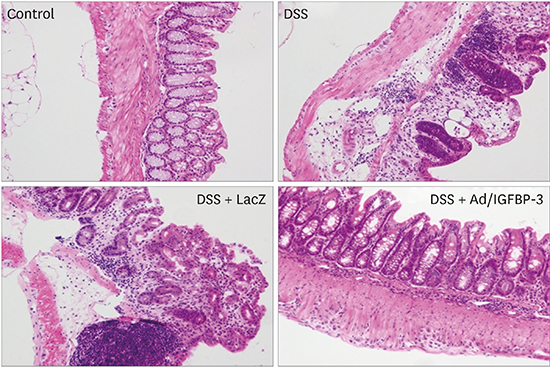
Paraffin-embedded colon tissues were sliced and stained. Immunohistochemical staining was performed with IGFBP-3 antibodies from Bioworld Technology. IGFBP-3 expression was compared among the 4 experimental groups. DSS with Ad/IGFBP-3 treated group showed greater IGFBP-3 expression than was observed in the other 3 groups (Fig. 6). The tissues were stained with hematoxylin-eosin for microscopic examination. In the control group, the mucosa and the submucosa showed normal crypts and goblet cells without thickening, edema, or infiltration of inflammatory cells. The colitis mucosa presented crypt distortion or absence, infiltration of inflammatory cells, edema, and erosions or ulceration in histological studies. Treatment with Ad/IGFBP-3 markedly reduced these inflammatory injuries, showing relatively preserved crypts (Fig. 7A-D). In the crypt and the involvement scoring system, the Ad/IGFBP-3 therapy group showed significantly lower damage scores compared to scores of other colitis groups (Fig. 7E, P < 0.05).
Fig. 6
Immunohistochemical staining for IGFBP-3 (× 200). IGFBP-3 expression. Pretreated Ad/IGFBP-3 induced the expression of IGFBP-3 in colonic mucosal cells.
IGFBP-3 = insulin-like growth factor-binding protein-3, Ad/IGFBP-3 = adenoviral vector system expressing insulin-like growth factor-binding protein-3, DSS = dextran sodium sulfate.

Fig. 7
The histologic findings and score of colonic mucosa. (A-D) Ad/IGFBP-3 treatment prevented DSS-induced damage in histologic evaluation. (A) Control, (B) DSS, (C) DSS + Ad/LacZ, (D) DSS + Ad/IGFBP-3. (D) DSS + Ad/IGFBP-3 group shows sustained crypt structure compared to the other DSS injury groups (hematoxylin-eosin stain, × 200). (E) Histological scoring of the colitis was performed. The severity was quantified by two scoring systems. The crypt abnormality and the Involvement of inflammation were significantly reduced in the Ad/IGFBP-3-treated group compared with Ad/LacZ-treated group. Statistical mean of crypt score and involvement score. DSS (3.67 and 3.67), DSS with Ad/LacZ (3.50 and 3.33), DSS with Ad/IGFBP-3 (1.83 and 2.67). aP = 0.009, bP = 0.014, and cP = 0.056 were calculated by Mann-Whitney U test.
Ad/IGFBP-3 = adenoviral vector system expressing insulin-like growth factor-binding protein-3, DSS = dextran sodium sulfate.

DISCUSSION
The IGF system consists of the growth hormones IGF-I and II, receptor for I or II, IGFBPs (IGFBP-1, 2, 3, 4, 5, and 6), and IGFBP protease.3 IGFBP-3 is the binding protein and carrier of IGF-I. It is the most abundant form of IGFBPs, and it controls the bioavailability of IGF-1 by distributing and interacting with the IGF-I receptor. Other known functions of IGFBP-3 are inducing apoptosis and promoting or inhibiting cell growth.3 Recent studies have proposed IGFBP-3 to have critical roles in anti-inflammatory functions through IGF-I-dependent or -independent pathways. Most of the circulating IGF-I is bound to IGFBPs, with the free form of IGF-I in circulation constituting less than 1%. The IGF-dependent pathway refers to the inhibition of biological functions of IGF signaling by binding to IGFs with higher affinity than IGF-receptors.18 IGFBPs also have their own biological actions, independent of IGFs. Numerous evidences suggest the ligand-independent biologic functions of IGFBP-3 such as inhibiting cell growth (anti-tumor effects), inducing apoptosis or cell growth, inhibiting preadipocyte differentiation, and anti-inflammatory effects. The mechanisms of these functions are unknown. Some are attributed to NF-κB inhibition and the interaction between IGFBP-3 and nuclear receptors (retinoid X receptor-α and retinoic acid receptor).319 Previous studies reported that IGFBP-3 significantly inhibits TNF-α-induced NF-κB activity.20
IGFBP-3 overexpression has been possible due to the development of adenoviral gene therapy. Adenoviruses are good mediators and are highly efficient at infecting cells. Over 95% of target cells express the transgene introduced by the adenovirus.21 Introducing a recombinant IGFBP-3 adenoviral (Ad/IGFBP-3) vector system enabled experiments on the intra- and extracellular functions of recombinant IGFBP-3.14 Furthermore, expression of Ad/IGFBP-3 under control of the CMV promoter suppresses of non-small cell lung cancer cell growth in vitro and reduces tumor size in vivo.22 Overexpressing PTEN in gastric cancer cells and downregulating c-myb in leukemic cells, using an adenoviral gene transfer system, inhibited cancer cell proliferation and IGF expression, and up-regulation of IGFBP-3 expression.2324 Recently, Han et al.4 proposed that pretreatment with Ad/IGFBP-3 inhibits NF-κB activity and expression of IL-8, VEGF, ICAM-1, and VCAM-1 in a prostate cancer model. IGFBP-3 suppressed tumor growth, angiogenesis, metastasis, and invasion, and the NF-κB pathway through activation of caspase-3/7, and -8.
Patients with IBD, whose condition is poorly controlled with various current medicines, exhibit clinical courses of intractable symptoms or exacerbations. Furthermore, many drugs for adults may be harmful for the growth or the development of children. For these reasons, we need a new approach for treating IBD in children. Systemic corticosteroids are very effective first-line drugs for inflammation, but they have some adverse effects including glucose intolerance, hypertension, headache, cataract, dermatitis, osteopenia, increased risk of infection, and decreased linear growth. About 30% of the patients show steroid dependency and flare-up after dose reduction.25 Recently, TNF-α inhibitors (infliximab, adalimumab) have shown higher success in inducing and maintaining remission in intractable patients who were unresponsive to conventional therapy. TNF-α is a pro-inflammatory cytokine derived from monocytes. These days, drug concepts have changed from a step-up approach to a top-down approach. TNF-α inhibitors are used as first-line drugs in patients with moderate to severe step, fistulizing, or penetrating lesions.26
The NF-κB pathway induces transcription of genes involved in cell inflammation, growth, survival, and angiogenesis.27 NF-κB is an important regulator of proinflammatory gene expression. Pro-inflammatory cytokines synthesized by NF-κB mediators include IL-1β, TNF-α, COX-2, and IL-6 and 8.28 NF-κB activation is associated with diverse chronic inflammatory diseases in humans. Asthma is a chronic airway inflammatory disease. An increased NF-κB activity was observed as pro-inflammatory cytokines and chemokines are overexpressed in asthmatic bronchial epithelial cells and up-regulation of IGFBP-3 blocks NF-κB signaling.6 Helicobacter pylori-induced gastritis is associated with increased NF-κB signaling in epithelial cells, and the severity of gastritis correlates with the amount of NF-κB positive cells.29 There is a report of NF-κB signaling activity in rheumatoid arthritis. Lee et al.5 suggested that Ad/IGFBP-3 gene therapy inhibits TNF-α-induced apoptosis and NF-κB signaling. Similarly, IBD is also associated with increased NF-κB pathway activity in mucosal cells.30 Conventional drugs for IBD, such as steroids and 5-aminosalicylic acid, inhibit the NF-κB pathway. NF-κB-targeted therapy plays a key role in advanced therapeutic strategies for treating chronic inflammatory diseases. This study revealed that the expression of phospho-p38MAPK, phospho-ERK, and NF-κB is decreased in Ad/IGFBP-3-treated IEC-6 cells.
Some studies have examined the relationship between the IGF system and IBD. In children with Crohn's disease or ulcerative colitis, growth retardation is a particularly common problem. In particular, the time interval from the onset of disease to diagnosis correlates with the severity of growth failure.31 Growth hormones and IGF-I secreted from the liver directly stimulate the growth plate in the long bone to proliferate growth plate chondrocytes. Patients with active IBD have significantly lower levels of IGF-I and IGFBP-3 compared to those of control or IBD patients in remission, but the levels normalize after inflammation subsides.32 Eivindson et al.12 reported that normalized levels of total IGF-I and IGFBP-3 were found after treatment with infliximab, whereas the levels of free IGF-I remained suppressed. They also reported that the levels of total IGF-1 and IGFBP-3 were markedly normalized after steroid treatment, but free IGF-1 levels increased within the first month of treatment.10 Undernutrition and poor caloric intake may result in IGF-1 and IGFBP-3 loss. Protein losing enteropathy (PLE) may, thus, be the expected reason of IGFBP-3 loss, but some studies showed that the degree of PLE does not correlate with the level of IGFBP-3 loss.33 Given that severe colitis results in the downregulation of IGFBP-3, this study suggests that the up-regulation of IGFBP-3 would result in colitis control. In the murine colitis model, Han et al.34 showed that growth hormone administration reduces disease activity of mucosal inflammation and improves weight gain through upregulating SOCS3 or downregulating STAT3 activation. This suggested that modulating the IGF system, including IGFBP-3, may have beneficial effects on colitis.
Oxidative injury is a crucial factor in IBD deterioration. We showed that in LPS-treated IEC-6 cells, ROS formation was increased and SOD level was decreased. Oxidative stress damages lipids, DNA, and proteins. ROS are neutralized by their conversion into water by Cu/Zn or Mn SOD, both of which serve as enzymatic antioxidants. Pretreatment with Ad/IGFBP-3 reduced ROS formation and sustained the level of SOD in this study. Pro-inflammatory cytokines are essential signals in the immune system. Intestinal epithelial cells, dendritic cells, macrophages, and T cells secrete various cytokines, such as IL-1β and TNF-α that regulate the inflammation response. In particular, TNF-α has various effects in the intestinal mucosa; it is a pivotal inflammatory cytokine in the cascade. TNF-α is associated with diverse autoimmune diseases including psoriasis and rheumatoid arthritis, and it plays a key role in IBD pathogenesis. Anti-TNF-α antibodies (infliximab and adalimumab) neutralize this cytokine and interrupt the inflammatory pathway. We detected increased expression of pro-inflammatory cytokines (COX-2, IL-1β, and TNF-α) in LPS-treated IEC-6 cells. Upon overexpression of IGFBP-3, the expression of these cytokines was decreased.
To conclude, we suggest that Ad/IGFBP-3 gene therapy can improve cell viability, act as an antioxidant, block the NF-κB pathway, and inhibit the expression of pro-inflammatory cytokines. In a DSS-induced colitis murine model, Ad/IGFBP-3 gene therapy showed anti-inflammatory effects in both gross and histological findings. In summary, the Ad/IGFBP-3 gene therapy has potential as a new therapeutic protocol in patients with intractable IBD.
 XML Download
XML Download