

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Lennox-Gastaut syndrome (LGS) is an epileptic syndrome with manifold symptomatalogy. This electro-clinical syndrome is composed of the triad of mental retardation, multiple seizure types and characteristic generalized slow spike-wave complexes (SSWs).1 The seizure types include tonic, atonic, atypical absence, myoclonic, partial , and generalized convulsive seizures.2 The syndrome may be idiopathic, cryptogenic or symptomatic. In the latter category CNS infections, tuberous sclerosis, cerebral malformations, traumatic childbirth, asphyxia of the neonate, frontal lobe pathology, and craniocerebral trauma have all been implicated. In approximately one-third of patients with LGS a history of West syndrome as a harbinger of LGS can be elicited.3 LGS is found in 1 to 4 percent of patients with childhood encephalopathy. Prognosis is unfavourable in the majority of cases. The electroencephalogram (EEG) is essential for the diagnosis of this syndrome.
In this paper, we report on two patients with LGS who were found to have dysgenesis of corpus callosum. We discuss the role of corpus callosum in the pathogenesis of secondary bilateral synchrony (SBS) in the EEG of patients with LGS.
CASE
This 4-year-old male was admitted to the hospital in December of 2012 for a neurological evaluation. He presented with a history of developmental delay and seizures. At 8 months of age he was diagnosed with West syndrome. Screening for inborn errors of metabolism was negative. His karyotype was found to be 46XY.
The patient's prenatal, perinatal, and postnatal history was reported to be unremarkable. The parents were not consanguineous. There was no family history of developmental delay or seizures.
Since age 6 months the patient had been experiencing tonic, myoclonic, atonic, and generalized convulsive seizures often precipitated by lack of sleep, crying, and fever. After a trial of numerous anticonvulsants in various combinations the patient's seizures finally responded successfully to a combination of vigabatrin and topiramate.
A neurological evaluation at King Khaled Hospital in December of 2012 showed the patient's weight and height to be within the 50th percentile, and the head circumference to be within the 25th percentile. He had large floppy ears and excessively long eyelashes. There were 2 cafe-au-lait spots in the lower extremities. There was a kyphoscoliosis of thoraco-lumbar spine.The patient was unable to turn in bed, sit, stand, walk. He had generalized hypotonia and hyporeflexia. He was unable to talk, and had severe intellectual disability. An ophthalmology consultation revealed no abnormalities.
Case report 2
The patient is an 11-year-old female who was admitted to the hospital for a neurological evaluation and seizure management. The patient presented with a history of intellectual- developmental delay, and refractory seizures.
Detailed prenatal, perinatal, and post-natal history was not available. She had multiple seizure types including tonic, atonic, myoclonic, and generalized convulsive seizures. Her developmental milestones were all delayed. She had been unsuccessfully treated with a combination of valproic acid, topiramate, levetiracetam, and phenobarbital. Family history was significant in that her younger sister had epilepsy.
Neurological examination showed a severely-disabled and wheelchair-bound female who was unable to talk, nor was she able to follow simple commands. She had microcephaly and facial dysmorphia. She had bilateral optic atrophy but no evidence of chorioretinitis. She had spastic quadriplegia. Deep tendon reflexes were 1-2 plus bilaterally and symmetrically in the upper extremities, 1 plus at the knees and absent at the ankles. There was no Babinski sign.
The EEG showed diffuse background slowing associated with prolonged bursts of high voltage, rhythmic 2 to 2.5 per second spike-wave and polyspike-wave complexes occurring over the right hemisphere with limited projection to the left hemisphere. Intermittent sharp waves and sharp slow waves occurred in the left temporal region independently. Intermittent brief episodes of sudden background flattening occurred in all leads bilaterally and synchronously (electrodecremental seizures) (Fig. 3). A treatment plan was formulated to simplify the anticonvulsant therapy and substitute an alternate regimen consisting of vigabatrin and topiramate.
DISCUSSION
The corpus callosum (CC) is the largest midline structure of the brain. It begins to develop around the 10th to 11th week of pregnancy, and consists of over 200 million nerve fibres that connect the two hemispheres of the brain. It transfers and integrates motor, sensory, and cognitive information between the cerebral hemispheres. It continues to mature throughout pregnancy, and through childhood and adolescence. The rostrum and genu of corpus callosum connect the motor and premotor cortex, the anterior half of CC integrates the somatosensory cortex, the splenium connects the parietal and occipital cortex, the posterior two-thirds of CC and dorsal splenium connect the auditory regions and the limbic system while the splenium and the truncus integrate the visual cortex.
The association of cryptogenic epilepsy and callosal dysgenesis was originally described by Aicardi and his co-workers.4 The authors described an X-linked dominant syndrome primarily consisting of infantile spasms, chorioretinitis, and complete or partial absence of CC. In Aicardi syndrome infantile spasms almost never evolve into LGS. This has been attributed to disruption of inter-hemispheric connections due to absence of CC, causing a failure of secondary bilateral synchrony (SBS) to occur.
The "unilateral" presence of SSWs in our case 2 suggests that callosal hypoplasia is indeed associated with impaired physiology of CC. Furthermore, this case may represent a variant of LGS where mental retardation and multiple seizure types exist without the presence of SBS. In such instances, neuroimaging studies are justified to investigate the underlying pathology including callosal dysgenesis or asymmetrical brain pathology. In the latter category the hemisphere where SSWs are attenuated or absent may paradoxically represent the site of predominant pathological involvement (Hirachovy5).
The EEG of our patient 1 did not show the typical SSWs of LGS. Instead, it showed generalized, bilaterally synchronous "notched delta", a poorly-defined pattern that has not been systematically studied in the EEG literature. However, the association of notched delta and LGS has been reported.6 Theoretically, notched delta may signify evolution of SSW in some cases of LGS. It is not clear whether the occurrence of this pattern in our patient reflected the effect of anticonvulsants on the EEG, transforming SSWs to notched delta. At this time, there is no scientific proof to support this hypothesis.
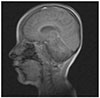
The mechanism of SBS in LGS is poorly-understood. However, it is hypothesized that SBS emanates from a focal ictal discharge, mainly frontal, which abruptly spreads to both hemispheres via the CC.78 The presence of SBS in our patient 1, whose MRI showed a preserved "genu", supports this theory. Accordingly, one might speculate that an alternative to the traditional partial or complete callosotomy would be resection of the genu only, to treat refractory generalized seizures. Further studies will of course be necessary to prove this point. In addition to anterior CC, the thalamic reticular cells may also play a pivotal role in the genesis of SBS in LGS8). Moreover, according to some investigators, genetics may also play a part in the occurrence of SBS.6
In our case 2, despite global callosal hypoplasia, the electrodecremental discharges occurred in all leads bilaterally and synchronously (Fig. 3). We hypothesize that these discharges emanate not from the cortex but from brain stem structures, in particular, the mesencephalon. Therefore, as previously suspected,9 in LGS a "dysfunctional" mesencephalon potentially generates pacemakers that project abnormal signals to the cortex via thalamic nuclei, resulting in the emergence of electrodecremental seizures.
A review of the literature reveals the significance of CC in integrating and synchronizing various inter-hemispheric activities. In a patient with Sanjad-Sakati syndrome associated with agenesis of the splenium of corpus callosum but intact genu, the inter-hemispheric synchrony of physiological background rhythms was maintained.10 In a case of ictal "periodic lateralized epileptiform discharges (PLEDs) associated with agenesis of CC there was no projection of the PLEDs to the contralateral hemisphere, indicating an inter-hemispheric dissociation.
The presence of hypoplasia of CC in our patients reflects a degenerative (destructive) rather than genetic insult to the brain, having occurred after the formation of CC at 12 weeks of gestational age, perhaps between 13 to 16 weeks of pregnancy. It is reasonable to assume that the LGS insult also occurred around that time. Indeed, a causal relationship has been theorized between agenesis of CC and other CNS anomalies. Future studies on this topic should focus on the timing and nature of LGS insult during pregnancy both in genetic and degenerative cases.
Our data suggest that hypoplasia of the genu of CC can interfere with the genesis of SBS in LGS, resulting in the appearance of unilateral or asymmetrical SSW (LGS variant). Furthermore, this callosal anomaly does not prevent the occurrence of electrodecremental seizures in LGS, suggesting a mesencephalic generator for the emergence of this EEG event.


 XML Download
XML Download