

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor
Genomic structural variants often display variable expressivity and penetrance. Variable expressivity can occur even within a single family, complicating genetic counseling [1]. For example, 1q21.1 structural variants are associated with variable phenotypes, including microcephaly, congenital heart malformations, autistic spectrum disorders, and developmental delay [1]. The main challenge is to link rare changes in large copy number detected by microarrays to a broad spectrum of phenotypic consequences and determine their clinical significance. The rationale of comparing our case with the previously reported cases is to explain why the subtle phenotypic differences occur from the same copy number variants.
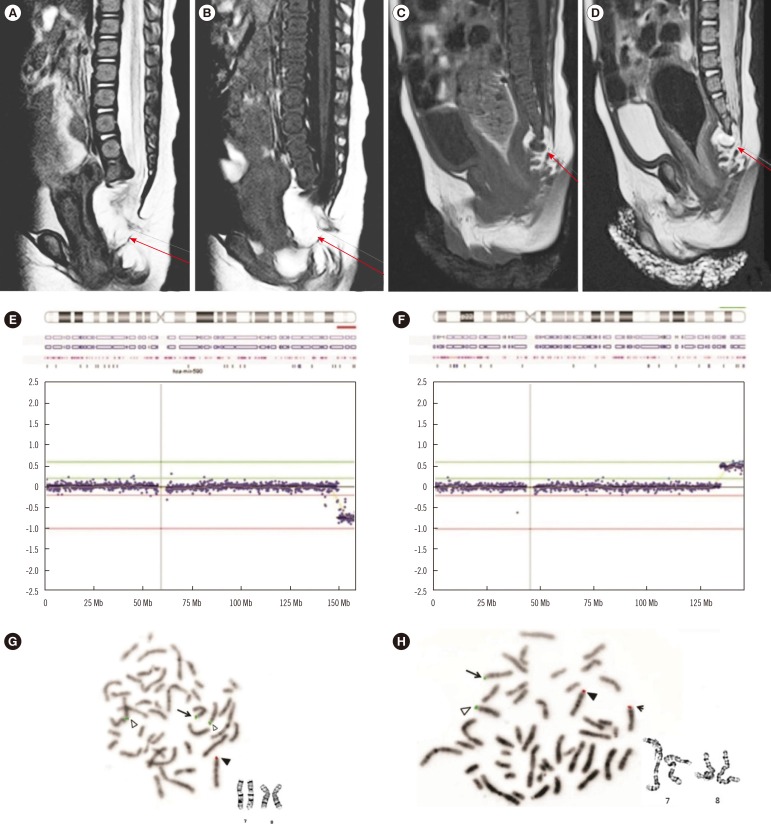
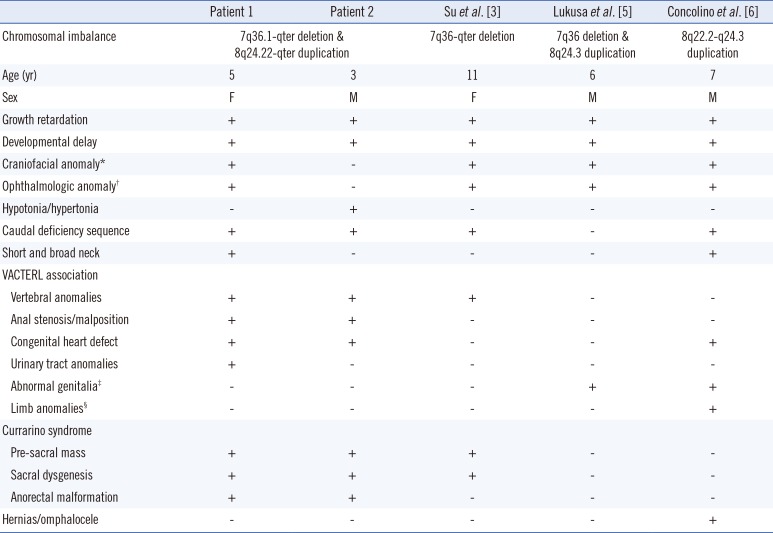
A 31-year-old mother was a healthy, G3P1-0-1-1 multiparous woman. Her first child was normal. The second female child (patient 1) was delivered vaginally at 38 weeks and 5 days gestation from non-consanguineous parents. Her head circumference, weight, and height at birth were 30 cm (<3rd percentile), 2,360g (<3rd percentile), and 44 cm (<3rd percentile), respectively, indicative of intrauterine growth restriction. Pertinent abnormalities included sacral dysgenesis, an imperforate anus, an atrial septal defect, and lipomyelomeningocele (Fig. 1A, B). Small eyes with poor fixation, ptosis, chorioretinal atrophy, and coloboma were accompanied by a duplicated kidney with hydroureteronephrosis on the right side (Table 1). Patient 1 underwent a sigmoid colostomy on day 5 and Pena operation at 4 months for an imperforate anus. The lipomyelomeningocele was repaired at 2 months of age, followed by the resection of a newly developing pre-sacral cystic mass at 19 months. This was confirmed as mature teratoma by biopsy. Her growth was delayed at the age of three years. The Bayley Scales of Infant Development (BSID) (II) at 2 yr of age showed mental and motor developmental ages of 6-7 months and 7 months, respectively.
Three years later, the mother became pregnant and underwent cordocentesis at 19.6 weeks because of the genetic abnormality in the previous child. She gave birth to a boy (patient 2) with a gestational age of 38 weeks and 5 days by vaginal delivery. The head circumference, weight, and height at birth were 34 cm (50-75th percentile), 2,635g (10-25th percentile), and 44 cm (<3rd percentile), respectively. Pertinent abnormalities included an atrial septal defect, pelvic mass, sacral dysgenesis, stenosis of the rectum and sigmoid colon, and a tethered cord (Table 1, Fig. 1C, D). Patient 2 underwent removal of the sacral mass, confirmed as mature teratoma by biopsy, and lipomyelomeningocele repair at 13 months of age. His growth was delayed at the age of 3 yr. BSID (II) at 2 yr showed a mental and motor developmental age of 6 months.
Patient 1's karyotype exhibited an apparently normal 46, XX. However, array-comparative genomic hybridization (CGH) using a high-resolution NimbleGen CGH 135K array (Roche NimbleGen, Madison, WI, USA) showed a 9.7-Mb terminal 7q deletion and a 12.0-Mb terminal 8q gain (Fig. 1E, F). FISH studies using the BAC clone RP11-354K9 on 7q36.3, and RP11-91F24 on 8q24.3, showed der(7)t(7;8)(q36.1;q24.22) in patient 1 (Fig. 1G), and a balanced translocation, t(7;8)(q36.1;q24.22) in the mother (Fig. 1H). This resulted in cytogenetic nomenclature of 46,XX,der(7)t(7;8)(q36.1;q24.22)mat.ish der(7)t(7;8)(RP11-354K9-,RP11-91F24+).arr[hg19]7q36.1q36.3 (149,479,066-159,128,663)×1, 8q24.22q24.3(134,220,817-146,304,022)×3 in patient 1. Patient 2 showed identical abnormal FISH results.
In our patients, the deleted region, 7q36.1-qter, contained 11 morbid OMIM genes (KCNH2, NOS3, ASB10, PRKAG2, DPP6, PAXIP1, SHH, LMBR1, MNX1, DNAJB6, WDR60). The duplicated region, 8q24.22-qter, contained 5 morbid OMIM genes (NDRG1, ZFAT1, KCNK9, TRAPPC9, PLEC1). The large changes in copy number encompassing many functional genes may have contributed to the phenotypic variability by changing the expression of dosage-sensitive genes, modifying gene regulation, exposing recessive alleles, and other mechanisms [2].
Both patients had phenotypic expressions of VACTERL (vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities) association and the Currarino syndrome showing subtle differences in severity (Table 1). These are associated with mutations in motor neuron and pancreas homeobox1 (MNX1) [3]. The pre-sacral mass (Fig. 1C, D) and sacral dysgenesis are suggestive of a ventral midline defect involving SHH and HLXB9 deletion [3]. In our patients, the atrial septal defect may be caused by 8q24.22-qter duplication near the critical region for cardiac septum formation [4]. The limb anomalies, abnormal genitalia, and hernia/omphalocele, known features of 7q36-qter deletion [5], or 8q22.2-q24.2 duplication [6], were not observed (Table 1).
There were several remarks to point out on the different phenotypes between the two siblings. PAXIP1 deletion may affect PAX2 function, contributing to coloboma and the renal anomaly seen in patient 1 [7]. The chromosomal deletion involving SHH with low penetrant variations may have caused the different ocular phenotypes in our two siblings [8]. Copy number variations (deletion or duplication) involving the imprinted domains in the vicinity of another imprinted gene, PEG1/MEST on 7q31-34 may restrict the placental growth [9] and contribute to the intrauterine growth restriction seen in patient 1. The epigenetic or genetic interactions along the locus between 7q-ter deletion and 8q-ter duplication may have caused changes in the regulation of imprinting domains. Collectively, the clinical significance may be involved with silenced imprinting domain caused by 7q36.1-q36.3 deletion involving PAXIP1 related PAX2 function and MNX1 [3579], with unbalanced duplications of 8q24.3 including KCNK9 [10].
This case adds to the literature that deletion at 7q36.1-qter, and duplication at 8q24.22-qter, may result in variable phenotypes from the same changes in copy number.
 XML Download
XML Download