

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Asthma is characterized as eosinophilic airway inflammation and hyperresponsiveness [1, 2]. T helper 2 (Th2) cytokines such as interleukin (IL)-5-activate eosinophils [3-7] and enhance eosinophil adhesion to vascular epithelial cells [8], and migration of eosinophils causes eosinophilic airway inflammation.
Interferon (IFN)-alpha has been widely used in the treatment of chronic hepatitis C [9-12]. IFN-alpha also favors both induction of Th1 cytokines and suppression of Th2 cytokines by signaling mechanisms [13-15]. IFN-alpha inhibits the production of IL-5 by CD4+ T cells [16] and inhibits eosinophils and CD4+ T cell recruitment into tissue [17]. In fact, the efficacy of therapeutic administration of IFN-alpha to patients with Churg-Strauss syndrome has been reported [18-20]. Consequently, these findings suggest that IFN-alpha may downregulate eosinophil activation and have an effect on severe asthma. To evaluate the effect of IFN-alpha for asthma, a guinea pig model of asthma was used for airway hyperresponsiveness, and human vascular endothelial cells and human eosinophils were used for the expression of adhesion molecules. IFN-alpha could be a viable treatment if its efficacy for asthma can be demonstrated, because IFN-alpha has been used widely for the treatment of chronic hepatitis C and there is ample information on IFN-alpha, including its side effects.
MATERIALS AND METHODS
Reagents
Human IFN-alpha (Hayashihara Lab, Osaka, Japan) composed of subtypes alpha 2, alpha 7, and alpha 8 was used for in vitro and in vivo experiments. Recombinant human (rh) IL-1 beta (Genzyme Co., MA, USA) and platelet activating factor (PAF; Sigma-Aldrich Co., MO, USA) were also used.
Antibodies
Anti-macrophage antigen-1 (Mac-1; CD11b, and mouse IgG2a) monoclonal antibody (mAb) and mouse isotype control mAbs (IgG1 and IgG2a) were purchased from Becton-Dickinson (CA, USA). Anti-human intercellular adhesion molecule-1 (ICAM-1; mouse IgG1) mAb and anti-human vascular adhesion molecule-1 (VCAM-1; mouse IgG1) were purchased from Genzyme Co. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse F(ab)'2 fragments were also purchased from Becton-Dickinson.
Sensitization
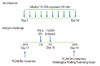
Guinea pigs were sensitized by exposure to aerosolized 10 mg/mL of ovalbumin (OVA; Sigma-Aldrich Co.) saline solution once a day for 10 consecutive days [21]. The aerosol was generated by an ultrasonic nebulizer (NE-IOB; Omuron Co., Ltd., Kyoto, Japan) applied to the center of a round tub. Guinea pigs were placed radially with their heads directed towards the aerosol-supplying center of the tub and were each exposed to an equivalent density of OVA aerosols for 10 min on each occasion. Seven days after the end of the 10-day exposure of OVA aerosols, and 3 h before the OVA challenge, the guinea pigs were intraperitoneally (IP) injected with IFN-alpha, which were suspended 2.0 mL/animal of (a) high-(2 × 107 U/kg) or (b) low dose- (2 × 106 U/kg) or (c) saline solution alone. Then, they were exposed to 10 mg/mL aerosolized OVA saline solution for 10 min using a DeVilbiss 646 nebulizer (DeVilbiss Co., PA, USA) driven by compressed air at 5 L/min. Airway hyperresponsiveness was measured immediately before and 24 h after OVA challenge (Fig. 1).
Measurement of airway hyperresponsiveness
Respiratory resistance (Rrs) was measured by a forced-oscillation technique (Mead's oscillation method) [22]. These measurements were carried out in inhalation-sensitized guinea pigs [23]. Conscious animals were placed in a body plethysmograph, leaving their heads outside and their necks restrained by a doughnut-shaped rubber balloon which scaled the body chamber, and an 18 Hz sine-wave oscillation was applied to their body surfaces. Oscillating pressure was generated (RC oscillator ORC 11; Kikusui Electronics Co., Kanagawa, Japan) with a 24 cm woofer speaker (Pioneer Co., Kanagawa, Japan) derived by a sine-wave generator and power amplifier (M-50; NEC Co., Tokyo, Japan). A rubber mask with a 200 mesh screen was snugly applied to the face in order to measure the flow rate. The flow rate through the mask was measured by detecting the pressure difference across the 200 mesh screen using a differential pressure transducer (DP-45-18; Validyne Engineering Co., CA, USA). Box pressure was measured by a pressure transducer (DP-45-26; Validyne Engineering Co.). The signals of the flow rate and the box pressure were amplified (Carrier Amplifier; Shizume Medical Co., Tokyo, Japan) and were then recorded on a polygraph recorder (WTR 751; Graphtec Co., Kanagawa, Japan), as well as on the X and Y axis of an oscilloscope (COS5021TM; Kikusui Electronics Co.). Rrs was calculated from the ratio of amplitude of the oscillating box pressure and flow rate fluctuations.
Airway responsiveness to inhaled histamine (Sigma-Aldrich Co.) was expressed using PC200-Rrs-histamine. Serially doubling dilutions of histamine solution of 39-20,000 µg/mL were prepared. Animals were exposed to the aerosol of each histamine solution for 10 min by increasing the concentration of each solution as described above. Rrs was measured immediately after inhalation, which was stopped when Rrs exceeded 200% of the baseline level. PC200-Rrs-histamine expressed the concentration of histamine solution to produce 100% increase in Rrs compared with baseline.
Eosinophil count
Animals were killed by IP injection of pentobarbital (100 mg/kg body weight). Trachea were removed and fixed in 10% formalin and embedded in paraffin, and 3-µm-thick sections were stained with May-Giemsa and Hansel solution. Epithelial and under basement membrane eosinophils were counted separately. The above protocol, including sensitization and measurement of airway hyperresponsiveness, was approved by the Committee of Laboratory Animal Research Center at Dokkyo Medical University (No. GP-9832).
Isolation of human eosinophils
Human eosinophils were isolated by the technique described by Tenor et al. [24]. Purity of attained eosinophils was > 99.5%, as assessed by Kimura staining after this procedure.
Adhesion assay
Human umbilical vein endothelial cells (HUVECs; Kurabou Biomedical, Osaka, Japan) were grown to confluence in a 24-well plate (Becton-Dickinson, Lincoln Park, NJ), and purified human eosinophils (2 × 105 cells/well) were added. Monolayers of HUVECs were pretreated with IFN-alpha or human albumin at 37℃ in a humidified CO2 (5%) incubator for 30 min and stimulated with 5 nM/mL of rh IL-1 beta for 4 h. After incubation, non-adherent eosinophils were collected, and the percentage of adhesion was calculated as follows: [(total cell number - non-adherent cell number)/total cell number] × 100.
Transendothelial migration assay
A Transendothelial migration assay was prepared using a modified version of Ebisawa's method [25]. HUVECs at 2 × 104 cells from the third passage were cultured on 2% gelatin-coated Transwell culture inserts (6.5-mm diameter polycarbonate membrane with 5-µm pores, Costar, MA, USA). Fresh endothelial cell growth medium (0.2 mL EBM; Sankojunyaku, Tokyo, Japan) was added to the upper and lower transwell chambers. A monolayer of confluent HUVECs formed within 7 days. To confirm HUVEC confluency, monolayers were stained routinely with Diff-Quik (American Scientific Products, McGraw Park, IL) before experiments. After preincubation for 30 min with IFN-alpha, the HUVECs monolayer was incubated at 37℃ for 4 h with 5 ng/mL of IL-1 beta or EBM only without glucocorticoid (control). After incubation, the culture medium with IFN-alpha and IL-1 beta was removed and rinsed twice. Then, 0.2 mg eosinophil suspension (2 × 106 cells/mL) was added to each upper chamber and incubated for 2 h at 37℃. After incubation, non-adherent cells in the upper chamber were removed. Eosinophils that had migrated and fallen into the lower chamber were removed to centrifugal tubes and centrifuged at 300 g for 5 min at 4℃. Migrated eosinophils were stained with trypan blue and counted.
Stimulation of eosinophils and HUVECs
Eosinophils resuspended in RPMI-1640 supplemented with 1% fetal bovine serum at a cell concentration of 1 × 106/mL were incubated for 30 min with IFN-alpha and added platelet-activating factor (PAF; 1 × 10-5 M/mL) for 15 min. HUVECs were detached from the culture flasks using 0.025% trypsin and 0.01% EDTA solution and were counted and resuspended at 1 × 106 cells/mL in EBM without glucocorticoid. After preincubation for 30 min with IFN-alpha, HUVECs were incubated with 5 ng/mL of rh IL-1 beta for 4 h. Cells were then washed twice with phosphate buffered saline and kept on ice until analyzed.
Immunofluorescence
Eosinophils were evaluated by immunofluorescence and flow cytometry for expression of CD11b, LFA-1a, and VLA4 before and after PAF stimulation. In addition, HUVECs were evaluated for expression of ICAM-1. Cells were labeled with saturating amounts of each mAb (~10-50 µL), and volume was made up to 100 µL where necessary with ice-cold flow cytometry buffer. After incubation for 30 min at 4℃, the cells were washed once with 2 mL of cold flow cytometry buffer, resuspended in PBS, and kept on ice until analyzed. When unlabeled mAbs were used, cells were mixed with an excess of FITC-conjugated F(ab')2 fragments of goat antibody to mouse immunoglobulins. After incubation for 15 min at 4℃, the cells were washed, resuspended in PBS, and kept on ice until analyzed.
Flow cytometry
Flow cytometry was performed on a FACScan instrument (Becton-Dickinson) equipped with an electronic (Coulter) volume sensor, which was standardized with fluorescent microbeads. The fluorescence intensity of 5,000 cells was determined from each sample by logarithmic amplification, which was converted to the linear equivalent by means of a Hewlett-Packard Consort 30 computer. Cell populations and debris were defined by gating the volume and 90° light scatter parameters. Dead cells were excluded from analysis by gating out propidium iodide-positive cells. The mean fluorescence of the population was calculated using the computer, and the value for the control antibody was subtracted from the value for the specific mAb to give a specific mean fluorescence value.
Statistical analysis
All statistical analyses were performed using Microsoft Excel®, StatView®, or JMP® for the t-test and Wilcoxon signed rank test. Differences with a p value < 0.05 were considered significant. Results are expressed as mean ± SEM values. PC200-Rrs-histamine values were converted into a logarithm for statistical calculation.
RESULTS
Effect of IFN-alpha on OVA-induced eosinophil infiltration into the trachea and on airway hyperresponsiveness
High- (2 × 107 U/kg) and low- (2 × 106 U/kg) dose IFN-alpha significantly inhibited eosinophil accumulation (1132 ± 70/mm2 and 1305 ± 68/mm2, respectively) compared with no treatment (1501 ± 64/mm2) (p < 0.01 and p < 0.05, respectively) (Fig. 2). Similarly, high- and low-dose IFN-alpha significantly decreased airway hyperresponsiveness (Rrs: 5000 ± 945 µg/mL histamine and Rrs: 1228 ± 365 µg/mL histamine, respectively) compared with no treatment (Rrs: 279 ± 68 µg/mL histamine) (p < 0.01, p < 0.05, respectively). High-dose IFN-alpha significantly decreased airway hyperresponsiveness compared with models before OVA challenge (1651 ± 325 µg/mL, respectively) (p < 0.05) (Fig. 3). Histological examination and measurement of airway hyperresponsiveness were performed separately, because histamine might induce airway inflammation.
Effects of IFN-alpha on adhesion molecule expression on human eosinophils and HUVECs
In vitro, IFN-alpha was suspended in culture medium at 1 × 105 U/mL (high-dose), 1 × 104 U/mL (middle-dose), or 1 × 103 U/mL (low-dose), and no treatment was used as a control. Mean fluorescence of ICAM-1 expression on HUVACs activated by 5 ng/mL of IL-1 beta was 221.7 ± 15.0, 237.8 ± 14.5, and 271.7 ± 28.7 for high-dose, middle-dose, low-dose pretreatment, respectively, compared with 279.3 ± 15.1 for no treatment (Fig. 4A). ICAM-1 expression was inhibited dose-dependently by IFN-alpha (p < 0.01, p < 0.05). Mean fluorescence of VCAM-1 expression on HUVACs activated by 5 ng/mL of IL-1 beta was 189.0 ± 29.7, 266.9 ± 23.0, and 256.5 ± 22.8 for high-dose, middle-dose, low-dose pretreatment, respectively, compared with 283.4 ± 30.9 for no treatment (Fig. 4B). VCAM-1 expression was inhibited by high-dose IFN-alpha only (p < 0.01). However, IFN-alpha did not suppress PAF-induced Mac-1 expression on human eosinophils (data not shown).
Effects of IFN-alpha on eosinophil adhesion and transmigration through HUVECs
For the adhesion investigation, the percent of adherent eosinophils was 52.6 ± 4.1%, 63.4 ± 4.1%, and 71.6 ± 4.1% for high-dose, middle-dose, low-dose pretreatment, respectively, compared with 83.8 ± 4.9% for no treatment (Fig. 5). IFN-alpha significantly and dose dependently inhibited eosinophil adhesion to HUVECs activated by 5 ng/mL of IL-1 beta (p < 0.01). Furthermore, the percent of migrated eosinophils was 6.7 ± 1.0%, 8.0 ± 1.3%, and 8.5 ± 1.4% for high-dose, middle-dose, and low-dose pretreatment, respectively, compared with 10.3 ± 1.3% for no treatment (Fig. 6). High dose IFN-alpha significantly inhibited eosinophil migration through IL-1 beta-activated HUVECs compared with controls (p < 0.02).
DISCUSSION
We found that IFN-alpha inhibited both airway eosinophilia and hyperresponsiveness in a guinea pig model of asthma. IFN-alpha inhibited the expression of ICAM-1 on vascular endothelial cells and the adherence of eosinophils to vascular endothelial cells. IFN-alpha also inhibited migration of eosinophils via vascular endothelial cells.
Several studies that evaluated the relationship between IFN-alpha and pulmonary function have been published and are in agreement with the present findings. For example, for severe corticosteroid-dependent asthma without chronic hepatitis C, IFN-alpha improved pulmonary function, including forced expiratory volume in 1 second (FEV1) and peak expiratory flow (PEF) [26-28]. On the other hand, patients with chronic hepatitis C did not show the same results. It was reported that two patients with both asthma and chronic hepatitis C experienced exacerbation of asthma promptly after the use of IFN-alpha [29]. In non-asthmatic patients with chronic hepatitis C, 3 of 17 experienced enhanced airway hyperresponsiveness after IFN-alpha treatment, although other pulmonary functions, including FEV1, showed no change between before and after treatment in any patients [30]. Another publication reported that the IFN response to chronic hepatitis C was associated with impaired responses to inhaled corticosteroid therapy in patients with both asthma and chronic hepatitis C, but no responders showed improved FEV1 by inhaled corticosteroid therapy [31]. Although IFN-alpha improves asthma in patients without chronic hepatitis C, we have to understand that IFN-alpha may cause exacerbation of asthma in patients with chronic hepatitis C.
In our study, low-dose IFN-alpha inhibited the expression of ICAM-1 but not VCAM-1. However, high-dose IFN-alpha inhibited expression of both ICAM-1and VCAM-1. Several studies have reported that treatment of IFN-alpha decreased the serum levels of soluble ICAM-1, but not soluble VCAM-1, in patients with chronic hepatitis C [32, 33]. Such results suggest that the main target of adhesion molecules will be ICAM-1, which can be inhibited by low-dose IFN-alpha. ICAM-1 expression on human endothelial cells is enhanced by IL-1 beta and tumor necrosis factor [34]. IFN-alpha inhibits ICAM-1 expression, because IFN-alpha inhibits IL-1 induced by endothelial cells [35]. Accordingly, eosinophil migration into tissue is inhibited.
In our study, the same results were shown both in vivo and in vitro, indicating that ICAM-1 may play an important role in airway eosinophilic inflammation. On the other hand, ICAM-1 expression is not always inhibited by IFN-alpha therapy. It was reported that 10 of 23 patients with metastatic malignant melanoma showed high expression of ICAM-1 after IFN-alpha therapy [36]. In addition, patients with metastatic malignant melanoma who developed depression had higher soluble ICAM-1 levels than non-depressed patients after treatment with IFN-alpha, compared with baseline levels [37]. IFN-alpha treatment may also contribute to neurotoxic effects. Furthermore, ICAM-1 expression in malignant diseases is increased and other signaling pathways in malignant diseases will be constructed to be increased ICAM-1 expression by IFN-alpha treatment.
IFN-alpha dose-dependently reduced the number of infiltrating eosinophils in airway tissue in our study. Two processes are candidate mechanisms through which IFN-alpha inhibits airway eosinophilic inflammation. The first process is inhibition of eosinophils by IFN-alpha via downregulation of T helper type 2 (Th2) cells. Here, IFN-alpha and IL-12 jointly promote differentiation of T helper type 0 (Th0) toward the T helper type 1 (Th1) phenotype, and IFN-alpha selectively stimulates Th1 cells to release IFN-gamma [38-40]. The activated Th1 cells downregulate Th2 cells by Th1-Th2 balance and inhibit the release of cytokines, which in turn activates eosinophils and mast cells, such as IL-4 and IL-5, from Th2 cells. Airway eosinophilic inflammation is subsequently inhibited by downregulation of Th2 cells. The second process is inhibition of the migration of eosinophils into airway tissue by IFN-alpha via ICAM-1 on HUVECs. The expression of HUVEC surface ICAM-1 is induced by tumor necrosis factor-alpha (TNF-alpha) and IL-1 [41], and the expression of ICAM-1 stimulated by IL-1 was inhibited by IFN-alpha in our study. The experiment for signaling pathways has not been performed, and we cannot explain the reason. Because ICAM-1 expression in malignant diseases is increased by IFN-alpha treatment, signaling pathways may not be simple. But, in an animal model of asthma without malignant diseases, IFN-alpha inhibits the expression of ICAM-1 on HUVECs. Thus, it is thought that IFN-alpha inhibits airway eosinophilic inflammation via both of these two processes, leading to inhibition of airway hyperresponsiveness.
In conclusion, IFN-alpha inhibits airway eosinophilic inflammation and airway hyperresponsiveness possibly by inhibition of ICAM-1 expression on endothelial cells. IFN-alpha is used widely for patients with chronic hepatitis C and can be used safely. Therefore, IFN-alpha treatment should be considered for uncontrolled patients with glucocorticoid-dependent severe asthma. However, when patients have malignant diseases or chronic virus infections, such as chronic hepatitis C, caution must be taken when using IFN-alpha because pulmonary function or airway hyperresponsiveness may be decreased when coexisting with those diseases.





 XML Download
XML Download