

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Asthma is a chronic inflammatory and allergic disease of the respiratory tract.1 In asthma patients, persistent inflammation and smooth muscle hyperplasia cause airway thickening and narrowing.2 When severe, asthma attacks lead to contraction of tracheal smooth muscle, which, in turn, causes difficulty breathing, coughing, and shortness of breath. In addition, the tracheal epithelial cells secrete more mucus, leading to airway obstruction and even suffocation and death.3
Mucins are large glycosylated proteins that form a major component of mucus, which is critical for the maintenance of the essential physiological functions of the airways.4 A subpopulation of bronchial epithelial cells, known as goblet cells, secrete mucus to protect the airways. A highly hydrated mucus gel functions together with the cilia of epithelial cells to capture dust and other respiratory pathogens, forming sputum. This process reduces the likelihood of lower respiratory tract infections.5 However, when inflammatory mediators stimulate epithelial cells of the airways to induce mucus hypersecretion, the airway can become obstructed and respiratory function can be reduced, particularly in patients with chronic airway diseases (e.g., asthma, chronic obstructive pulmonary disease, and cystic fibrosis).5,6
Overproduction of mucus can obstruct the airway, which can result in difficulty breathing and death for asthma patients.7 Patients with acute asthma symptoms are, therefore, administered drugs to stimulate expansion of the trachea, and to reduce goblet cell inflammation and mucus secretion. β2-agonists are commonly used to expand the trachea during asthma attacks.8,9 However, recent studies have found that long-term inhalation of β2-agonists leads to the development of drug tolerance, requiring the patient to inhale an increased dose of drug in order to suppress acute asthma symptoms.8 There is, therefore, a need to identify viable therapeutic alternatives to current asthma medications.
Th2 activity can exacerbate asthma progression.3 Activated Th2 cells secrete more interleukin (IL)-4, IL-5, IL-9, and IL-13, causing airway hyperresponsiveness (AHR), eosinophil infiltration, goblet cell hyperplasia, and excessive mucus secretion.10 Furthermore, Th2 cells induce secretion of IgE from B-cells, and stimulate mast cells to release additional allergic and inflammatory factors.11 These characteristics make Th2 cells an enticing target for amelioration of asthma symptoms.
Lovastatin, an HMG-CoA reductase inhibitor, is a member of the family of statins that can be isolated from red yeast rice.12 In clinical trials, statin has proved effective in improving the symptoms of high blood cholesterol.13 Recent studies have found that statins also have anti-inflammatory effects.14 Specifically, simvastatin and lovastatin can improve inflammatory symptoms in murine model of asthma.15,16 It is, therefore, worthwhile to investigate the potential of statins as a novel treatment for asthma. This study aimed to evaluate whether lovastatin improves bronchial mucus plugging, lung inflammation, and cytokine gene expression in murine model of asthma. In parallel, we investigated the effect of lovastatin on inflammatory tracheal epithelial cells in vitro.
MATERIALS AND METHODS
Animals
Six- to eight-week-old female BALB/c mice, weighing ~20 g, each were purchased from the National Laboratory Animal Center in Taipei, Taiwan. Animal experimental protocols and procedures were approved by the Laboratory Animal Care Committee of Chang Gung University of Science and Technology and Chang Gung University.
Sensitization, airway challenge, and drug treatment
Murine model of asthma were induced as described previously.17 Briefly, the mice were sensitized by intraperitoneal injection of ovalbumin (OVA) (Sigma, St. Louis, MO, USA) with aluminum hydroxide (Thermo, Rockford, IL, USA), in 200-µL normal saline, on days 1-3 and 14. A nebulizer (DeVilbiss Pulmo-Aide 5650D, USA) was also used for inhalation administration of 2% OVA to challenged mice for 30 minutes on days 14, 17, 20, 23, and 27. Normal saline or lovastatin was administered orally to mice on days 14-27.
Mice were randomly divided into four groups (12-14 animals per group): (1) mice sensitized and challenged with normal saline (N); (2) mice sensitized and challenged with OVA (OVA), (3) OVA-sensitized mice orally administered 10 mg/kg lovastatin (L10); (4) OVA-sensitized mice orally administered 40 mg/kg lovastatin (L40).
Measurement and analysis of AHR
On day 28, a whole-body plethysmograph chamber (Buxco Electronics, Troy, NY, USA) was used to assess AHR, and evaluate airway function after aerosolization with methacholine, as described previously.18 Briefly, mice inhaled increasing doses of methacholine (0-40 mg/mL) for 3 minutes, and enhanced pause (Penh) values were recorded.
Cell counting and collection of bronchoalveolar lavage fluid
On day 29, mice were anesthetized and sacrificed for bronchoalveolar lavage fluid (BALF) collection, as described previously.18 After intubation of the trachea, the lungs were washed three times with 1-mL normal saline. Cells were stained with Liu's stain solution to determine the eosinophil percentage and perform a cell count.
Histological analysis of lung tissue
Lungs were harvested and fixed in 10% formalin, paraffinized, cut into sections of 6-µm thickness, and mounted onto glass slides. To assay eosinophil infiltration, slides were stained with hematoxylin and eosin (H&E). To assess lung epithelial goblet cell hyperplasia, slides were subjected to periodic acid-Schiff (PAS) staining (Sigma), as described previously.19
Immunohistochemistry
Lung tissue slides were de-waxed and incubated with mucin 5AC (MUC5AC) antibody (1:50; Santa Cruz, CA, USA) overnight, as described previously.19 Samples were then washed and incubated with secondary antibody (HRP-conjugated goat anti-rabbit antibody) for 30 minutes. Immunolabeling was visualized using 3,3'-diaminobenzidine (DAB) substrate, and the staining reaction was observed with a light microscope.
Lung RNA isolation and real-time PCR for gene expression
Total ribonucleic acid (RNA) was isolated from lung tissue using TRIzol reagent (Life Technologies, Carlsbad, CA, USA). Complementary DNA (cDNA) was synthesized using a cDNA Synthesis kit (Life Technologies) and l-µg RNA per sample. Complementary DNA gene expression was assayed by real-time PCR performed with a spectrofluorometric thermal cycler (iCycler; Bio-Rad Laboratories, Hercules, CA, USA) and the SYBR Green system (Fermentas, Thermo, Waltham, MA, USA). Specific primers were designed as follows: murine CCL11 (forward 5'-CCA TTG TGT TCC TCA ATA ATC C-3', reverse 5'-GGC TTC ATG TAG TTC CAG AT-3'), CCL24 (forward 5'-AGG CAG TGA GAA CCA AGT-3', reverse 5'-GCG TCA ATA CCT ATG TCC AA-3'), intercellular adhesion molecule 1 (ICAM-1; forward 5'-AGA CGC AGA GGA CCT TAA-3', reverse 5'-CAC ACT TCA CAG TTA CTT GG-3'), Muc5AC (forward 5'-CTG TTA CTA TGC GAT GTG TAG-3', reverse 5'-GTG GCG TGG TAG ATG TAG-3'), IFN-γ (forward 5'-TGA GAC AAT GAA CGC TAC A-3', reverse 5'-CCA CAT CTA TGC CAC TTG A-3'), IL-6 (forward 5'-CCG CTA TGA AGT TCC TCT C-3', reverse 5'-GGT ATC CTC TGT GAA GTC TC-3'), IL-4 (forward 5'-CGT GCT TGA AGA AGA ACT C-3', reverse 5'-TGT GGA CTT GGA CTC ATT C-3'), IL-5 (forward 5'-GGT AAT GTA GCC AAG GAT AAC-3', reverse 5'-TCC ATC TCC AGC ACT TCA-3'), IL-13 (forward 5'-GCA GCA TGG TAT GGA GTG-3', reverse 5'-GGT CCT GTA GAT GGC ATT G-3').
Serum collection and splenocyte cultures
Blood samples were centrifuged at 6,000 rpm for 5 minutes at 4℃ to obtain serum for collection of OVA-specific antibodies, as described previously.20 Splenocytes (5×106 cells/mL) were cultured in RPMI 1640 Medium (Life Technologies) supplemented with 100 µg/mL OVA for 5 continuous days. Supernatants were collected for assessment of cytokine concentrations.
Enzyme-linked immunosorbent assay
Cell culture supernatants were assayed using enzyme-linked immunosorbent assay (ELISA) kits for IL-4, IL-5, IL-6, IL-13, CCL11, CCL24, and ICAM-1 (R&D Systems, Minneapolis, MN, USA), as described previously.19,21 Serum OVA-specific antibodies, IgG1, IgG2a, and IgE, were measured using a specific ELISA kit (BD Biosciences). Sera from OVA-sensitized mice were pooled and used to create OVA-IgG1 and OVA-IgG2a standard curves for quantification of OVA-specific IgG1 and OVA-IgG2a based on OD450 readings. OVA-specific IgE levels were assayed by diluting serum fivefold and measuring the OD450.
Culture and lovastatin treatment of BEAS-2B cells
Human bronchial epithelial cells (BEAS-2B cell line) were seeded onto 24-well plates and maintained in DMEM/F12 medium (Life Technologies). After pretreatment with lovastatin (10, 40 µM) for 1 hour, the cultures were supplemented with 10 ng/mL tumor necrosis factor (TNF)-α and 10 ng/mL IL-4 for 48 hours, as described previously.17 The supernatants were collected, and cytokine or chemokine levels were assayed using ELISA kits.
Cell-cell adhesion assay
After lovastatin treatment, BEAS-2B cells were incubated with 10 ng/mL TNF-α for 24 hours, as described previously.19 HL-60 cells were treated with 0.5 mM butyric acid for 7 days, to stimulate differentiation into eosinophil-like cells, as described previously.22 HL-60 cells were then treated with calcein AM solution (Sigma) and co-cultured with BEAS-2B cells for 1 hour. After washing the cells, a fluorescence microscope (Olympus, Tokyo, Japan) was use to observe adhesion of HL-60 cells to BEAS-2B cells.
RESULTS
Effect of lovastatin on goblet cell hyperplasia and lung expression of MUC5AC
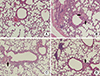
The lungs from sacrificed animals were fixed and stained with PAS to observe goblet cell hyperplasia (Fig. 1). Increased goblet cell hyperplasia was observed in the tracheas of OVA-sensitized mice, compared to control mice. OVA-sensitized mice treated with 10 or 40 mg/kg lovastatin exhibited significantly reduced goblet cell hyperplasia, compared to OVA-sensitized mice that were not treated with lovastatin.
Immunohistochemical staining revealed that expression of MUC5AC protein was elevated in OVA-sensitized mice, compared to normal mice (Fig. 2). However, MUC5AC expression was significantly inhibited in OVA-sensitized mice treated with lovastatin, compared to OVA-sensitized mice that were not treated with lovastatin. Thus, OVA sensitized mice that were orally administered with lovastatin exhibited significantly less goblet cell hyperplasia and MUC5AC mucin expression, resulting in decreased airway obstruction.
Effect of lovastatin on cytokine and chemokine expression in the lung
To investigate the effect of lovastatin on the expression of MUC5AC, cytokines, and chemokines in the lungs of OVA sensitized mice, real-time PCR was used to assess gene expression levels. As shown in Fig. 3, lovastatin significantly increased the expression of the genes encoding CCL11, CCL24, MUC5AC, ICAM-1, IL-4, IL-5, IL-6, and IL-13 in OVA-sensitized mice. However, the OVA-induced increases in gene expression were significantly suppressed by treatment with 40 mg/kg lovastatin, which also caused upregulation of IFN-γ expression.
Effect of lovastatin on lung eosinophil infiltration
Eotaxins induce migration of eosinophils into the lungs.3 Real-time PCR analysis showed that lovastatin decreased expression of eotaxins, including CCL11 and CCL24. Hematoxylin and eosin (H&E) staining of lung sections revealed that OVA-sensitized mice exhibit more eosinophil infiltration between the trachea and the blood vessels than normal control mice (Fig. 4). This infiltration was significantly suppressed by lovastatin treatment.
Effect of lovastatin on inflammatory mediators in activated BEAS-2B cells
To elucidate the role of lovastatin in the suppression of the release of inflammatory mediators from bronchial epithelial cells, BEAS-2B cells were stimulated with IL-4 and TNF-α. Lovastatin significantly reduced levels of IL-6, ICAM-1, and eotaxins (CCL11 and CCL24) in stimulated BEAS-2B cells (Fig. 5).
Inhibition of eosinophil adhesion to TNF-α-activated BEAS-2B cells by lovastatin
In the present study, we found that lovastatin decreased the expression of ICAM-1 and eotaxins, proteins that promote adhesion of eosinophils to inflammatory BEAS-2B cells. Next, we investigated whether lovastatin could suppress the adhesion of eosinophil-like HL-60 cells to inflammatory BEAS-2B cells (Fig. 6). Calcein AM was used to visualize cell-cell adhesion. TNF-α-stimulation resulted in increased adhesion of HL-60 cells to BEAS-2B cultures. Pretreatment with lovastatin, however, inhibited this adhesion significantly.
Lovastatin reduces eosinophil numbers in BALF
The number of eosinophils and total cells were measured in BALF collected from sacrificed mice (Fig. 7A and B). Both the total cell count and the number of eosinophils were significantly reduced in lovastatin-treated OVA-sensitized mice, compared with the OVA group (total cell numbers: L10, 1.06×106±1.26×105/mL, P<0.05; L40, 9.26×105±1.59×105/mL, P<0.01 vs OVA, 1.47×106±2.88×105/mL; eosinophil numbers: L10, 6.19×105±1.73×105/mL, P<0.05; L40, 4.83×105±1.16×105/mL, P <0.01 vs OVA, 9.24×105±2.93×105/mL). Administration of lovastatin to OVA-sensitized mice resulted in a significant decrease in the ratio of eosinophils to total cells, when compared with the OVA group (L10, 48.3±6.6% vs 67.2.3±11.1%, P<0.01; L40, 41.1±9.1% vs 67.2.3±11.1%, P <0.01).
Lovastatin improves AHR in OVA-sensitized mice
On day 28, the Penh value was recorded, as a measure of AHR at 24 hours after aerosol inhalation of the final OVA challenge (Fig. 7C). Mice inhaled various doses of aerosolized methacholine (0-40 mg/mL), before being placed into a plethysmograph chamber for recording of the Penh value. At 40 mg/mL methacholine, the Penh value was 6.99±1.12 in OVA-sensitized mice. Treatment with lovastatin significantly suppressed Penh values in the L10 and L40 groups, compared to the OVA group (L10, 4.69±0.65, P<0.01; L40, 3.06±0.68, P<0.01). These results indicate that lovastatin has a significant ability to decrease AHR in OVA-sensitized mice.
Effect of lovastatin on cytokine levels in splenocyte culture supernatant and OVA-specific antibodies in serum
In serum, treatment with 40 mg/kg, but not 10 mg/kg, lovastatin significantly reduced OVA-IgE and increased OVA-IgG2a, but did not change OVA-IgG1 levels (Fig. 8A, B, and C). In addition, lovastatin did not change cholesterol levels in serum (Fig. 8D). In splenocyte culture supernatant, treatment with 40 mg/kg lovastatin significantly reduced IL-4, IL-5, and IL-13 levels, compared to the OVA group (Fig. 8E, F, and G).
DISCUSSION
Asthma is an allergic inflammatory disease of the airways.1 In patients with chronic asthma, airway smooth muscle proliferation can cause airway thickening and narrowing.2 During acute asthma attacks, the surrounding smooth muscle shrinks the airway, and goblet cells of the trachea increase mucus secretion, leading to airway obstruction, coughing, shortness of breath, or suffocation.23 Reducing goblet cell hyperplasia is predicted to reduce airway obstruction caused by excessive mucus secretion.
Mucins are a major class of glycoproteins produced by epithelial cells.6,24 Approximately 20 mucin genes have been identified in humans, and more than 12 of these are expressed in the respiratory tract.4 In healthy individuals, goblet cells express primarily MUC5AC protein to facilitate clearance of toxic molecules and microbes from the airway by cilia.5,24 However, a previous study found that overexpression of mucin obstructs the airways in asthmatics, as a result of allergic stimulation.23 In the present study, we found that lovastatin suppresses goblet cell hyperplasia in the airways of murine model of asthma. Immunohistochemical staining showed that MUC5AC expression is significantly decreased in lung tissue, and real-time PCR analysis indicated that MUC5AC gene expression is significantly inhibited by lovastatin. We can conclude that lovastatin significantly inhibits airway mucus secretion, which may reduce suffocation and death in murine model of asthma.
When the tracheal epithelial cells of asthmatic patients make contact with allergens, an inflammatory response is triggered.23 Inflammatory epithelial cells secrete more mucus and release many inflammatory cytokines and chemokines.24,25 We found that lovastatin significantly inhibits gene expression of CCL11, and CCL24 in activated tracheal epithelial cells, and IL-6, CCL11, and CCL24 in the lungs of OVA challenged mice. CCL11 and CCL24 are chemokines that induce eosinophil migration into inflammatory tissue. Consistent with this, we found that lovastatin significantly inhibited eosinophil infiltration in the lungs of murine model of asthma.
Statins have been shown to inhibit HMG-CoA reductase and block cholesterol synthesis in the liver.12 Recent studies have found that statins also improve endothelial function, and prevent coronary artery thrombus formation in patients.15 Statins may even decrease the prevalence of stroke and heart attacks when there is no history of cardiovascular disease.14 Several studies demonstrate that statins improve the inflammatory profile of pneumonia in patients, by blocking Rho signaling.16 The anti-inflammatory effects of statins are associated with a decrease in IL-6 and C-reactive protein levels in asthmatic patients. IL-6 is a pro-inflammatory cytokine that causes lung injury and fibrosis.13 We show here that statins inhibit IL-6 activity; lovastatin reduces IL-6 secretion, thereby helping to reduce damage to the respiratory tract or lung fibrosis.
Eosinophils secrete major basic protein, eosinophil cationic protein, eosinophil peroxidase, and eosinophil-derived neurotoxin, to cause tissue inflammation and damage to the lung tissue.26 Hence, treatment with lovastatin to suppress eosinophil infiltration into the lungs is likely to reduce airway inflammation in asthmatic patients. In addition, inflammatory epithelial cells of the airway express ICAM-1 to facilitate eosinophil adhesion and subsequent migration into the airways.24 Using real-time PCR, we confirmed that lovastatin can inhibit ICAM-1 gene expression in lung tissue. We also found that inflammatory tracheal epithelial cells significantly express ICAM-1, but that this expression was reduced by lovastatin treatment. To confirm that lovastatin suppresses the ability of eosinophils to adhere to inflammatory airway epithelium, eosinophil-like HL-60 cells were applied to BEAS-2B cells activated in vitro. In this assay system, lovastatin significantly inhibited cell-cell adhesion, allowing us to conclude that lovastatin can reduce eosinophil infiltration into lung tissue. We suggest that lovastatin significantly reduces the inflammatory response of airway epithelial cells, leading to reduced eosinophil infiltration into lung tissue and improvement of asthma symptoms.
Previous experiments demonstrated that lovastatin inhibits AHR in allergic asthma model of rats.27 In our experimental murine model of asthma, we found that lovastatin also significantly inhibited AHR, eosinophil infiltration, goblet cell hyperplasia, and mucus hypersecretion in murine model of asthma. Asthma is considered a Th2-polarizing cytokine disease.3 We therefore examined the expression of IFN-γ, a Th1-polarizing cytokine. Lovastatin increased IFN-γ gene expression in the lungs, but not in spleen cell supernatants (data not show). Lovastatin also suppressed the expression of Th2-associated cytokines in lung tissue. These data suggest that lovastatin has significant immunomodulatory effects in the lungs of murine model of allergic asthma. Previous experiments confirmed that Th2-associated cytokines are induced during the development of asthma.11 IL-4 enhances B-cell activation and induces class-switching, leading to an increase in the levels of IgE and IgG1.28 Lovastatin decreased OVA-specific IgE levels in the serum of murine model of asthma, but did not inhibit OVA-IgG1 production. IgE induces mast cell activation, leading to allergy and inflammation.29 Importantly, IL-5 stimulates bone marrow cells to differentiate into eosinophils.30 In addition, tracheal epithelial cells secrete eotaxins (CCL11 and CCL24) to induce eosinophil migration into inflammatory lung tissue.24 We found that lovastatin decreases the levels of IL-5 in splenocytes and lung tissue, but did not confirm whether eosinophil numbers were decreased in the circulatory system. However, lovastatin did significantly decrease eosinophil infiltration into lung tissue and BALF. We believe that lovastatin reduces airway inflammation by inhibiting expression of eotaxins and ICAM-1, which, in turn, reduces the migration of eosinophils into lung tissue.
IL-13 enhances AHR in murine model of asthma.3 A previous study found that asthma was induced in the absence of AHR in IL-13-knockout mice.31 Administration of IL-13 to wild-type murine model of asthma enhances AHR.32 Surprisingly, IL-13 gene expression was 102-fold higher in OVA-sensitized mice than in normal mice. Treatment with 40 mg/kg lovastatin significantly inhibited IL-13 gene expression (27-fold decrease compared with OVA-sensitized mice). These data suggest that the improvement of AHR in murine model of asthma treated with lovastatin is caused by inhibition of IL-13 expression.
Previous studies have confirmed that simvastatin, lovastatin, and other statin drugs have anti-inflammatory effects that attenuate asthma symptoms in murine model of asthma.33,34 Simvastatin and rosuvastatin can modulate the levels of IL-6 and TNF-α in BALF, suppress mucus secretion, and ameliorate airway remodeling.13,35 In another study, pravastatin was shown to suppress airway inflammation by inhibiting IL-17 production.36 Pravastatin and pitavastatin have also been demonstrated to exert anti-inflammatory effects, which are associated with suppressed inflammatory cytokine production, via a mechanism involving inhibition of RhoA activation. Simvastatin and lovastatin can reduce Rho expression in the lungs of murine model of asthma, and can reduce cholesterol levels in serum.16,27 However, lovastatin did not inhibit the levels of Rho proteins in lung tissue in our experimental model (data not shown). In addition, we observed no change in blood cholesterol levels in murine model of asthma or lovastatin-treated mice.
Our aim was to investigate whether lovastatin improves mucus secretion in the lungs of murine model of asthma. Our data demonstrate that lovastatin attenuates airway inflammation and mucus secretion by suppressing expression of eotaxins and Th2 cytokines in mice with OVA-induced asthma.







 XML Download
XML Download