

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hereditary angioedema (HAE) is a rare condition with an estimated incidence of 1/10,000 to 1/50,000 in Western countries. Uncontrolled activation of the classical complement pathway is the key mechanism of HAE.1 There are three reported types of HAE,2 whereas acquired angioedema is most frequently associated with lymphoproliferative and autoimmune disorders or with some medications such as ACE inhibitors and plasmin activators.3 Typically, patients with HAE present with recurrent severe angioedema of the face and upper and lower extremities together with abdominal pain and laryngeal edema, which is sometimes life threatening, and there is usually a family history.2-5 No gender or race predominance has been described.6
CASE REPORT
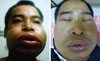
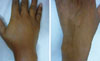
The patient, a 40-year-old man, had a 16-year history of recurrent weekly abdominal pain and swelling of the hands and lips without urticaria, in association with trauma or psychological and physical stress. He visited the emergency room after a day of hard work during which he developed severe swelling of the lips, around the eyes, and of the right hand (the left hand was normal), without urticaria or dyspnea. The symptoms had progressed gradually over time. He had not taken any medications for his symptoms. He denied a history of drug use, including use of anti-hypertensive agents. The patient had a brother who had died of recurrent facial and laryngeal swelling 5 years earlier. The physical examination revealed a normal heart rate with regular rhythm, normal oxygenation, and normal blood pressure. Diffuse swelling of his face, both lips, and right hand were noted (Figs. 1 and 2). He was treated with subcutaneous epinephrine and systemic antihistamine and corticosteroid. One day later, the patient was better and was discharged.
In view of the clinical and family histories and clinical presentation of this patient, we diagnosed HAE, despite a normal complement C4 level. We could not measure the C1 esterase inhibitor (C1-INH) level, as this test is not available in our country.
To prevent acute attacks of HAE, we started danazol 200 mg per day for 1 month, according to the Milan protocol for prophylaxis at the outpatient clinic,3,4,8-10 and observed no episodes of HAE during that month. We reduced the dose of danazol to 100 mg per day. There were no episodes of swelling and no adverse reactions for more than 2 months.
DISCUSSION
Landerman et al.11 characterized HAE in 1962. The role of C1-INH in regulating the contact system by inactivating both plasma kallikrein and factor XIIa was elucidated in the 1970s and 1980s.11,13
HAE has a strong family history associated with a quantitative (type I) or qualitative (type II) deficiency of C1-INH caused by mutations of the C1-INH gene.2 Type I HAE (85% of cases) presents with reduced C1-INH protein and function, while in type II (15% of cases), C1-INH has normal protein levels, but reduced function. Type III HAE, which presents with normal C1-INH protein and function, is an estrogen-dependent inherited form found mostly in females, with an otherwise unknown defect.2,13
The diagnosis of HAE is based on the typical clinical symptoms, family history, and laboratory findings such as lower C4, C1q, and C1-INH levels. Our patient had typical clinical findings with a positive family history, but a normal C4 level. Although this might have been a laboratory error, some cases of HAE with a normal C4 level have been reported.14 We could not measure the C1q or C1-INH level. In addition, the diagnosis of HAE can be based on a favorable clinical response to danazol treatment according to the Milan protocol for prophylaxis.3,4,8-10 Based on these findings, we postulate that our patient has type I HAE, even though he had a normal C4 level. He will require danazol therapy and long-term monitoring.3,4,10
Approaches to the diagnosis and management of HAE vary among countries, as a full laboratory setting and some treatments are not available in developing countries. Earlier treatments were developed empirically and include the fibrinolysis inhibitors epsilon aminocaproic acid and tranexamic acid and attenuated androgens such as danozol.3,4,10 In recent years, new agents such as C1 esterase inhibitor protein replacement products, a plasma kallikrein inhibitor, and a bradykinin receptor antagonist have been introduced.15,16
To prevent acute attacks, we used danazol, which is the only drug available in Vietnam, based on results using danazol in HAE prophylaxis.4,9,10,17 We prescribed danazol for 3 months and found that 100 mg of danazol daily prevented acute attacks of HAE in our patient.
In conclusion, we report the first probable case of HAE in Vietnam successfully treated with danazol. The diagnosis was based on the typical clinical symptoms, positive family history, and successful treatment with danazol. Further investigations are needed to identify more patients and define their clinical and laboratory findings in Vietnam.
 XML Download
XML Download