

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Exofocal postischemic neuronal death in the substantia nigra (SN) is a well-known example of anterograde transsynaptic cell death.1-3 Loss of inhibitory γ-aminobutyric acid (GABA) ergic afferent fibers from the neostriatum causes transsynaptic
cell degeneration in the SN.
The globus pallidus (GP) is divided into two segments: the external segment (GPe) and the internal segment (GPi). The GPi and the SN pars reticulata (SNr) have similar functional roles, and their differences merely reflect their topographical organization.4 Exofocal neuronal death could theoretically occur in the GPi after striatal injury, but this has not been reported previously. Herein we report two cases of delayed GPi injury after ischemic striatal injury.
Case Report
Case 1
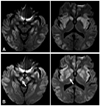
A 70-year-old woman visited the emergency room because of decreased mentality. She had been diagnosed with diabetes mellitus, and medication had been initiated 1 month prior to the visit. Two days before admission she was unable to recognize her family, and 1 day before admission she experienced drowsiness. On admission she exhibited a stuporous mental state. Her blood glucose level and oxygen tension were 169 mg/dL and 69.1 mm Hg, respectively. Cerebrospinal fluid findings were normal. Brain magnetic resonance imaging (MRI) was performed on the first day of admission; a diffusion-weighted image disclosed high-intensity signals in the caudate nucleus, putamen, and temporooccipital cortex on both sides. Follow-up (diffusion-weighted) MRI performed 10 days after admission revealed a decreased signal intensity in the caudate head and putamen on both sides, but new high-intensity signals had developed in the SN and GP on both sides (Fig. 1).
Case 2
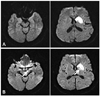
A 40-year-old man v isited the emergency room because of right-sided weakness. Medical Research Council grade 4 weakness was noted on the right side upon neurological examination. No sensory signs were observed. Diffusion-weighted MRI of the brain performed on the second day of admission revealed high-intensity signal lesions in the left caudate nucleus and putamen. Although there was no change in the symptoms, a follow-up MRI was performed 4 days after admission; this disclosed newly developed high-intensity signals in the left SN and GP (Fig. 2). The patient's weakness improved without development of any extrapyramidal symptoms.
Discussion
Pallidonigral damage after ischemia has been reported previously, but such reports are not supported by early MRI findings; therefore, the authors of those reports concluded that the pallidonigral damage was the result of the primary ischemic injury.5,6 In the cases described herein, both patients underwent both early and follow-up MRI, and changes in signal intensity in the SN and GP were seen only in later MRI scans. Therefore, we believe that the GP lesions were delayed changes, because the high-intensity signals in the GP and SN were not observed during the initial MRI. The timeline of the change in signal intensity in the GP was similar to that in the SN, and both can be attributed to exofocal postischemic neuronal death.
Exofocal postischemic neuronal death in the SN has been studied extensively in rat models; however, these studies have not revealed secondary changes to the rat GP, and therefore many investigators have inferred that GP damage in humans is primary in origin. In nonprimates the GPe is simply referred to as the GP.4 The nonprimate GPi is not equivalent to the primate GPi; it is small and is buried in the fibers of the internal capsule to form the entopeduncular nucleus. Thus, it is not surprising that exofocal neuronal damage cannot be observed in the rat GPi after striatal damage. Furthermore, the middle cerebral artery occlusion method was used to induce striatal damage in the rat model,1,2 which may have primarily damaged the GPi, thereby explaining why exofocal neuronal damage was not observed in the GPi.
Degeneration of the ipsilateral SN in humans following infarction in the striatum has been reported,3 but those authors did not report secondary changes in the GPi. However, all of the patients in that study underwent MRI only once, and hence the GPi injury might have been considered a primary infarction.
Injection of a GABAergic antagonist into the GPi in monkeys causes dyskinesia in the contralateral forelimb.7,8 Similarly, isolated GP lesions cause motor disturbances.9 However, no movement symptoms developed in our two cases; in case 1 we believe that this was due to the patient's stuporous mentality.
The exact mechanism underlying preservation of the GPe is unknown. One possible mechanism is that although both the GPe and GPi receive inhibitory inputs from the striatum, the GPi receives inputs from substance-P- and dynorphin-containing neurons (direct pathway), while the GPe receives both those direct-pathway neurons together with additional inputs from enkephalin-containing neurons (indirect pathway).10
We performed follow-up MRI despite unchanged symptoms in our two cases because we predicted that there would be changes to the MRI findings. Marked astrocyte swelling developed on day 4 following injury in rats.1 Thus, in our case 2, the follow-up MRI was performed 4 days after the symptom onset. In case 1, the follow-up MRI was delayed until 10 days after symptom onset because of the general condition of the patient. Exofocal anterograde transsynaptic neuronal death in the GP was more prominent in case 1. These observations suggest that exofocal neuronal death develops later in humans than in rats.
The GPi can be subjected to delayed damage via the mechanism of anterograde transsynaptic cell death. However, it is important that this is distinguished from recurrent ischemic injuries such as stroke.
 XML Download
XML Download