

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mucopolysaccharidosis II (MPS II, Hunter syndrome) is a lysosomal storage disorder; it is caused by a deficiency in the lysosomal enzyme iduronate-2-sulfatase (IdS), which is involved in the degradation of glycosaminoglycan (GAG).1 MPS II is an X-linked recessive disorder and occurs predominantly in males. The accumulation of GAG in tissues and organs is responsible for the associated clinical abnormalities.1 The liver is the primary site of accumulation of GAG; as a result, differenchepatomegaly is an invariable finding in patients with this disorder.1 Enzyme replacement therapy (ERT) with recombinant human IdS has been reported to be effective for the treatment of MPS II.2-5 An IdS-knockout (KO) mouse model was constructed to study the effects of ERT on mice with MPS II.
Recently, a patient with MPS II and hypoglycemia was treated; the patient had palpitations, was irritable, and had increased sweating and episodes of syncope. The reduced amount of glycogen in the hepatocytes was thought to cause accumulation of GAG, which might have been responsible for the observed hypoglycemia. This could be explained by the findings of our pilot study, where the liver pathology of IdS-KO mice showed a decreased amount of glycogen in the hepatocytes. To confirm these findings, the frequency of fasting hypoglycemia in patients with MPS II was investigated. In addition, changes in accumulation of glycogen and GAG in the hepatocytes of IdS-KO mice before and after recombinant IdS infusion was evaluated.
MATERIALS AND METHODS
Hypoglycemia after an 8-hour fast in patients with MPS II
Plasma glucose levels in 50 patients with MPS II were evaluated. Hypoglycemia was defined as a plasma glucose concentration of less than 55 mg/dL.6 The measurements were performed at 8 a.m. after an 8-hour fast. Glucose levels were also measured 2 hours after a meal.
IDS knockout mice
A KO mouse model was developed by Jin, DK, MD (Department
of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea) and was reported previously.7 Briefly, a murine model of Hunter syndrome was prepared by replacing part of the IdS gene, exon 2 and exon 3, with the neomycin resistance gene. Carrier females were bred with male mice with a B6/129 background strain, producing heterogeneous females and hemizygous male knock-out mice, as well as wild-type (WT) male and female littermates. The genotype of all mice used in these experiments was confirmed by polymerase chain reaction of DNA obtained from a tail snip.
Grouping
The animals were categorized into four groups. Eight normal mice were included in group 1. Group 2 consisted of 26410 IdS-KO mice without ERT. In group 3 there were four IdS-KO mice treated with 0.15 mg/kg of IdS, and in group 4, three IdS-KO mice treated by a 0.5 mg/kg IdS infusion.
Enzyme replacement therapy
The recombinant IdS enzymes used in this study were remnants of idursulfase (Elaprase®, Shire, Cambridge MA, USA) used for patient treatment. The remnants were used after informed consent was obtained from the parents of the patients. Recombinant IdS was injected into the lateral tail vein of mice in groups 3 and 4. ERT was initiated at 4 weeks of age, and continued every week until 20 weeks of age.
Pathology examinations for GAG and glycogen
At approximately 20 weeks of age, the mice were sacrificed and a comprehensive necropsy was performed. The livers were dissected and fixed in formalin, and made in paraffin block for light microscopic examinataion. The 4-6 µm thickness sections were put on glass slides and stained with periodic acid-Schiff (PAS) and digested periodic acid-Schiff (d-PAS), which stains tissue glycogen, and colloidal iron, which stains for excessive GAG. A routine pathology examination was performed in a blind manner by the pathologist. Under the light microscopic (LM) examination, semiquantitative verification of the GAG content of Kupffer cells was performed, scoring from absent (0) to global (2) and for the glycogen content of liver tissue, ranging from scanty (0.5) to global (3). The specimens were examined three times by the same pathologist.
Measurements of glycogen weights
The glycogen content of liver tissue was measured indirectly using amyloglucosidase digestion of the homogenates of liver tissue and weighing the amount of glucose present.
Electron microscopic examination
For the electron microscopic (EM) examination, the tissue was fixed in 2.5% glutaraldehyde solution, postfixed in 1.5% osmium tetroxide solution, dehydrated, and fixed firmly into polybed resin. Microthin section and staining with lead citrate and uranyl acetated were done. A comparison of the changes in the liver tissue before and after ERT was performed by EM examination.
Statistical analysis
A comparison of GAG and glycogen among the groups was analyzed by the Kruskal-Wallis test. To verify differences among groups, the results were reanalyzed using least significant difference (LSD) using ranks. Glycogen wet weight and histological semiquantitation were analyzed using the Spearman rank correlation coefficient.
RESULTS
Hypoglycemia after an 8-hour fast in MPS II patients
The mean glucose level after an 8-hour fast was 94.1 ± 23.7 mg/dL. Two (4%) out of 50 patients had fasting hypoglycemia. The mean plasma glucose level 2 hours after a meal was 110.9 ± 17.8 mg/dL. The levels were within normal range in all patients.
Pathological findings in the murine model
Upon routine histological examination, there was no significant difference in architecture or inflammatory changes before and after ERT. However, there were changes in GAG and glycogen content. GAG accumulation in liver tissues was compared among the groups, based on semiquantitation of colloidal iron staining. There were significant differences in the GAG assay among the groups (p = 0.015). Group 2 had a higher concentration of GAG compared to the other three groups, based on positive iron colloidal staining. Reduction of GAG content in the treated mice of groups 3 and 4 reflected the effects of ERT. However, there was no difference between groups 3 and 4 based on the dose of enzyme infusion or between group 1 and the groups treated with ERT (Table 1).
The glycogen content in the cytoplasm of hepatocytes, in the liver tissue of the mice, also differed among the four groups based on the findings of the semiquantitation periodic acid Schiff positive findings; the findings showed sensitivity to diastase digestion (p = 0.005). Group 1 had a higher concentration of glycogen than groups 2 and 3. Comparison between the two treated groups showed that the group with a higher dose of enzyme infusion had higher glycogen content than the group with a lower dose of enzyme infusion by the LSD (Table 2).
Glycogen weights
The fresh liver tissue from the mice was analyzed to determine glycogen content, and compared with the results of the semiquantitation pathology. There were significant differences in glycogen content among the four groups (p = 0.009)(Table 2). The mean glycogen content of group 1 was 5.75 ± 1.43 wet weight (%), which was more than those of group 2 [2.80 ± 1.07 wet weight (%)] and group 3 [2.80 ± 2.18 wet weight (%)]. The glycogen content for group 4 was 5.27 ± 1.92 wet weight (%). The glycogen wet weights were positively correlated with the results of the histological semiquantitation (p = 0.001, Spearman correlation coefficient = 0.74349)
Electron microscopic examination
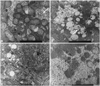
The ultrastructural findings observed on electron microscopy were consistent with the light microscopy findings for iron colloidal, PAS and d-PAS stains with regards to GAG and glycogen content. Group 2 showed accumulation of GAG in the lysosomes and scanty glycogen content in the cytoplasm. In group 3, the GAG in the lysosomes decreased, but restoration of glycogen in the cytoplasm was not remarkable. However, in group 4, GAG in the lysosomes had nearly disappeared and the glycogen particles in the cytoplasm were restored (Fig. 1).
DISCUSSION
Most of the patients with MPS II did not have hypoglycemia after an 8-hour fast. Even though two patients had fasting hypoglycemia, and one of them showed hypoglycemic symptoms, the fasting glucose levels in most patients were normal. This finding was contrary to the expectation that patients with MPS II would have hepatic glycogen depletion contributing to fasting hypoglycemia. Instead, these findings suggest that glucose metabolism, including hepatic glycogen synthesis and gluconeogenesis, were functioning well despite GAG accumulation and hypothetical glycogen depletion in the MPS II hepatocytes.
The findings from the murine model showed that GAG decreased in both the low and high dose ERT groups; however, glycogen was restored to normal range only in the high dose ERT group, which implies that high dose ERT is required to achieve the desired response. This result is consistent with the findings reported by Garcia, et al.,5 where urine GAG levels deceased in a dose-dependent manner. The reason why GAG semiquantitation assay results in this study did not show a significant difference between groups 3 and 4 might be due to the small number of samples in the two groups.
Resnick, et al.8 reported that the histological findings of liver biopsy samples from 27 MPS cases showed no architectural changes, inflammatory infiltrates, or extensive fibrosis. The results of this study also showed no specific histological changes in liver tissue, except for glycogen content depletion and GAG accumulation in the MPS II murine model.
The results of this study suggest that ERT in MPS II mice may influence hepatocyte glycogen content as well as lysosomal GAG. However, there is no known relationship between the regulation of synthesis, storage, and degradation of glycogen and the lysosomal accumulation and degradation of GAG in the liver. Therefore, the mechanism by which the amount of glycogen in the cytoplasm appears to be inversely proportional to the lysosomal GAG, based on the amount of enzyme supplementation, remains unclear. Two possible mechanisms have been proposed: 1) MPS II is associated with GAG accumulation in the lysosomes, resulting in lysosomal expansion. As GAG accumulates, space for normal glycogen in the cytoplasm is reduced. By contrast, after ERT, lysosomal GAG disappears and glycogen in the cytoplasm is built up to the level of normal hepatocytes. 2) Certain metabolites produced in MPS II may block the glycogen synthesis pathway, leading to glycogen depletion. After ERT, the metabolites may disappear, restoring normal glucose metabolism, which mediates stimulation of insulin release and glycogen synthesis.
Further studies linking hepatic metabolites produced in MPS II with glycogen metabolism may lead to better understanding of the mechanisms involved in hepatocyte glycogen depletion. The murine model did not provide evidence of hepatic fibrosis or cirrhosis. This finding is consistent with the results of Resnick, et al.,8 who reported fibrotic changes in liver biopsy samples from 27 infant MPS cases. Hepatic fibrosis in patients with MPS has been reported in the literature, mainly in adult cases.9,10 Parfrey and Hutchins suggested that hepatic fibrosis is due to abnormal accumulation of hepatotoxic metabolites.9 The MPS II murine model in this study showed that liver damage was not significant; lysosomal accumulation of GAG and histological changes during were observed in the early stages, but no fibrosis. Furthermore, histological changes returned to normal after ERT. Therefore, these findings suggest that ERT can be an effective treatment and should be started as early as possible during childhood.
In conclusion, high dose ERT in MPS II mice led to marked disappearance of lysosomal GAG and restoration of normal levels of glycogen in the cytoplasm of the hepatocytes. Normal accumulation of glycogen in hepatocytes is disturbed by the metabolites produced in this storage disease; however, this process is reversible.


 XML Download
XML Download