

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
World-wide population of obese people is growing fast [1]. Obesity is an important risk factor for hypertension [2]. Obese individuals have increased plasma level of renin-angiotensin-aldosterone system (RAAS) component including renin, angiotensinogen, angiotensin II (Ang II), and aldosterone [3]. RAAS plays a critical role in the elevation of blood pressure via regulating vascular contractility and blood volume [4]. High fat diet (HFD)-induced hypertension is established animal model to study renal and cardiovascular factors for the obesity-related hypertension [5678]. There are lines of evidence that inhibition of Ang II prevents HFD-induced hypertension [9].
Most forms of hypertension ultimately result from an increased vascular tone that leads to an elevated peripheral resistance [10]. Despite the clear role of Ang II in hypertension through increased vascular contraction [11] and increase in plasma level of Ang II by obesity [3], most studies on Ang II in obesity hypertension focused on water retention and blood volume expansion [12]. Thus far, a few studies on the mechanism of Ang II-induced vascular hyper-contractility in HFD-fed individuals have revealed that increased upstream mediators such as Ang II type 1 (AT1) receptor or angiotensin converting enzyme (ACE) are responsible for vascular smooth muscle (VSM) hypercontractility. Experiment using VSM cell model of metabolic syndrome demonstrated that increased release of reactive oxygen species (ROS) elevated Ang II-induced Ca2+ release and AT1 expression supporting augmented aortic contraction in response to Ang II [1314]. However, the clear role of increased basal level of Ang II in maintained resting VSM hyper-contractility in obese hypertensive individuals is not elucidated yet. Here, we studied the resting status of VSM contractile proteins affected by HFD and the mechanism underlying Ang II-induced hyper-contractility focusing on the alterations of VSM contractile proteins to elucidate the mechanism of HFD-induced hypertension.
VSM contraction absolutely depends on 20 KDa MLC phosphorylation that is determined by balance of MLC kinase (MLCK) and MLC phosphatase (MLCP) [15]. MLCK is activated by intracellular calcium and then phosphorylates MLC resulting in VSMC contraction. On the other hand MLCP is inhibited in calcium-independent manner that is occurred by phosphorylation of myosin phosphatase target subunit1 (MYPT1) at Thr853 residue or inhibition of catalytic subunit pp1c by C-kinase potentiated Protein phosphatase-1 Inhibitor (CPI17) resulting in increased MLC phosphorylation and VSM contraction [16]. Therefore altered amount and activation status of these contractile mediators by HFD should be critical for the obesity-induced hypertension and hyper-reactivity to Ang II. In this study, we tested the hypothesis that altered regulatory proteins for the vascular contraction such as MLCK and MLCP inhibitor CPI-17 are responsible for HFD-induced hyper-contractility and hypertension.
METHODS
Experimental animals
All animal experiments were conducted in accordance with guidelines of the National Institutes of Health for the care and use of laboratory animals. Experimental protocol (KM-2014-43) was approved by institutional Animal Care and Use Committee at Keimyung University. Eight week-old male Sprague-Dawley rats were purchased from Koatec (Daegu, Korea) and used. They were fed with 60% Kcal fat diet (TD.06414, Harlan Teklad, Madison, WI, USA) or its control diet containing 10% Kcal fat diet (TD.94048, Harlan Teklad, Madison, WI, USA) and had free access to water for 16 weeks as others induced diet-induced obesity previously [17].
Plasma lipid profiling
The plasma levels of total cholesterol, HDL-cholesterol, LDL cholesterol, triglycerides, and free fatty acids were measured by an automatic biochemical Analyzer (Roche Diagnostics, Basel, Switzerland).
Ang II enzyme immunoassay
Angiotensin II enzyme immunoassay kit (Cat. EK-002-12; Phoenix Pharmaceutical Inc., Burlingame, CA) was used for measuring the concentration of angiotensin II in rat plasma. Fifty microliter of rat plasma in duplicate was used for this assay. All the procedures were conducted according to the instruction of the assay kit.
Blood pressure measurement
Blood pressure (BP) of rats was measured by tail cuff method. Rats were preheated on a hot plate at 35℃ for 10 min and then placed in restrainer. A cuff with a pneumatic pulse sensor was attached to the tail. BP values were recorded on a CODA High Throughput Noninvasive Blood Pressure system (Kent Scientific, Torrington, CT, USA) with heating and were averaged from ten consecutive readings obtained from each rat.
Aortic ring preparation
To minimize the pain and stress, rats were anesthetized with pentobarbital sodium (50 mg/kg ip) before sacrifice. The thoracic aorta was excised from the rats under anesthesia. By excising aorta, rats were sacrificed. The excised aorta was placed into ice-cold modified Krebs-Henseleit buffer, pH 7.4. The aortae were cleaned of all adherent connective tissue and cut into rings (4 mm long). The endothelium was removed by gentle rubbing of the intimal surface using tip of small forceps.
Organ bath experiment
The aortic rings were horizontally mounted in 3 ml organ baths filled with Krebs-Henseleit buffer (pH 7.4), continuously bubbling with gas containing 95% O2 and 5% CO2. The changes in isometric tension of the rings were measured with a digital force isometric transducer (Grass, West Warwick, RI, USA) connected to data acquisition system (AD Instruments, New South Wales, Australia). The rings were equilibrated for 1 h under an initial tension of 2 g, with the buffer being changed every 10 min. After equilibration, the rings were challenged with 50 mM KCl and force was measured. After wash out and 1 h of equilibrium final concentration of 10 nM Ang II were added to the baths to obtain vascular contractile response. The contractions were induced at a basal tension of 2 g and calculated as grams tension per aortic ring.
Western blot analysis
Protein samples were prepared and western blot analysis were performed as previously described [18]. Protein samples were electrophoresed on 10~12% polyacrylamide gel with 0.1% SDS and transferred to PVDF membranes, and then subjected to an immunoblotting with antibodies against pMLC at Thr18 and Ser19 (Santa Cruz, CA, USA ), p-MYPT1 at Thr853 (Santa Cruz, CA, USA), p-CPI-17 at Thr38 (Millipore, Darmstadt, Germany), and MLCK (Santa Cruz, CA, USA). For the loading control anti-GAPDH antibody (Novus Biologicals, CO, USA) was used. Horseradish peroxidase-conjugated secondary antibodies (anti-rabbit: Santa Cruz, CA, USA; anti-goat: Bethyl, Montgomery, TX, USA; anti-mouse: Bethyl, Montgomery, TX, USA) were applied, and immunoblots were visualized using chemiluminescence reagent (PerkinElmer Life Sciences, MA, USA). Densities of immunoblots were quantified using image analysis software ImageJ (NIH, Bethesda, ML, USA).
RESULTS
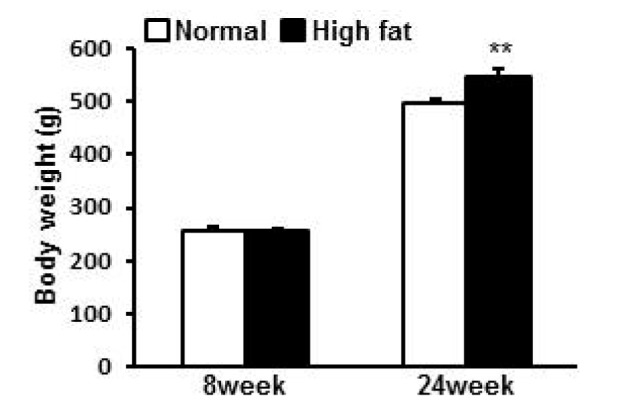
HFD accelerated body weight gaining
From 8 week of age, rats were randomly divided into 2 groups for normal diet (ND) or HFD. At 8 week old, there was no difference in the body weight between groups (258.2±4.4 g, 257.6±3.8 g for ND and HFD group respectively). With time, both ND group and HFD group gained weight. At 24 weeks, body weights were 498.2±6.5 g and 546.6±12.8 g for ND and HFD group respectively. ND group gained 240.0 g while HFD group gained 289.0 g showing greater weight gaining by 20% in HFD group compared to ND group (p=0.003) (Fig. 1).
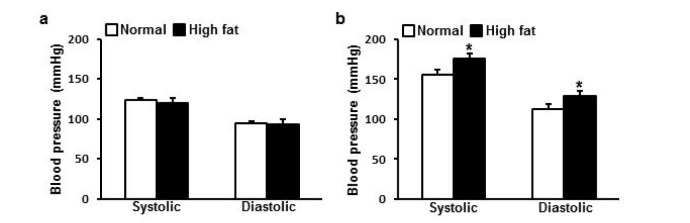
HFD accelerated BP increase
At starting point systolic BP was 123.2±2.9 mmHg and 120.2±5.8 mmHg, and diastolic BP was 94.2±2.8 mmHg and 93.1±6.8 mmHg for ND and HFD group respectively showing no difference between groups (Fig. 2a). With differential diet for 16 weeks, systolic BP and diastolic BP of both groups increased. Systolic BP was 155.1±6.9 mmHg and 176.0±7.0 mmHg, and diastolic BP was 112.9±6.6 mmHg and 128.7±6.3 mmHg for ND and HFD group respectively showing accelerated increase in HFD group compared to ND group (p=0.023, p=0.049 for systolic BP and diastolic BP respectively) (Fig. 2b).
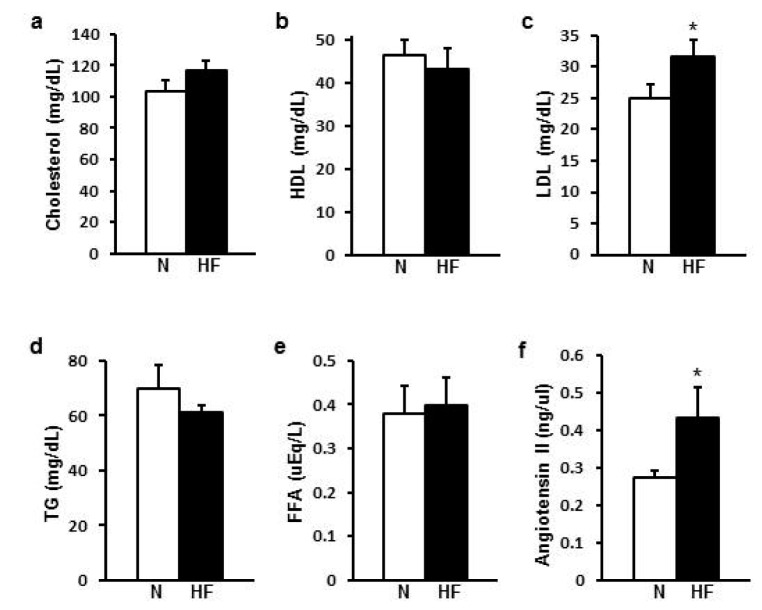
HFD increased plasma LDL and Ang II
For the plasma lipid profiling, LDL cholesterol was higher in HFD group than ND group (p=0.032) but other lipids such as HDL cholesterol, triglycerides, free fatty acids did not show any difference between groups (Fig. 3a to e). Plasma Ang II was higher in HFD group (0.432±0.082 ng/µl) than in ND group (0.275±0.016 ng/µl) (p=0.034) (Fig. 3f).
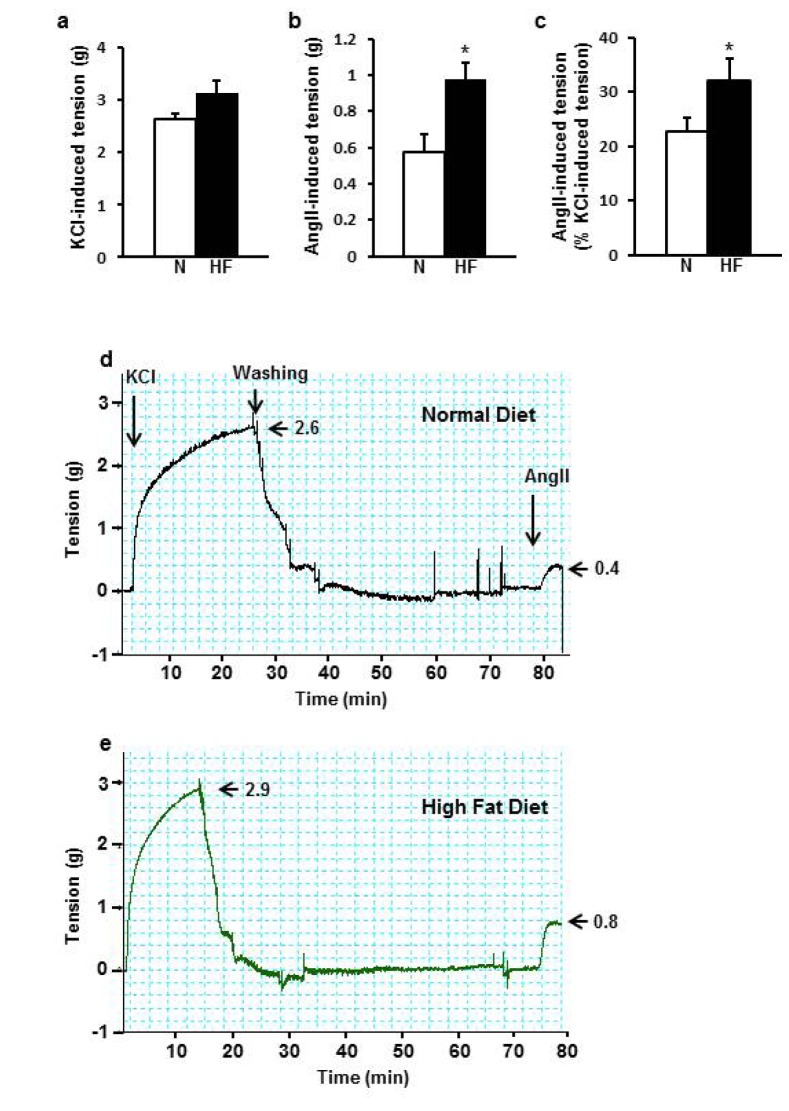
HFD enhanced vascular contractility in response to Ang II
In order to test whether HFD-induced hypertension is caused by increased Ang II-induced vascular contraction, I determined vascular contraction against Ang II normalized by KCl-induced contraction to minimize the difference in the initial viability between aorta rings. KCl-induced contraction was 2.6±0.1 g and 3.1±0.4 g (Fig. 4a). Ang II-induced contraction was 0.6±0.1 g in ND and 1.0±0.1 g in HFD group (Fig. 4b). Normalized Ang II-induced contraction was 22.7±2.5% in ND group and 32.1±4.1% in HFD group showing greater vascular contractility in HFD group (p=0.035) by 41.4% (Fig. 4c).
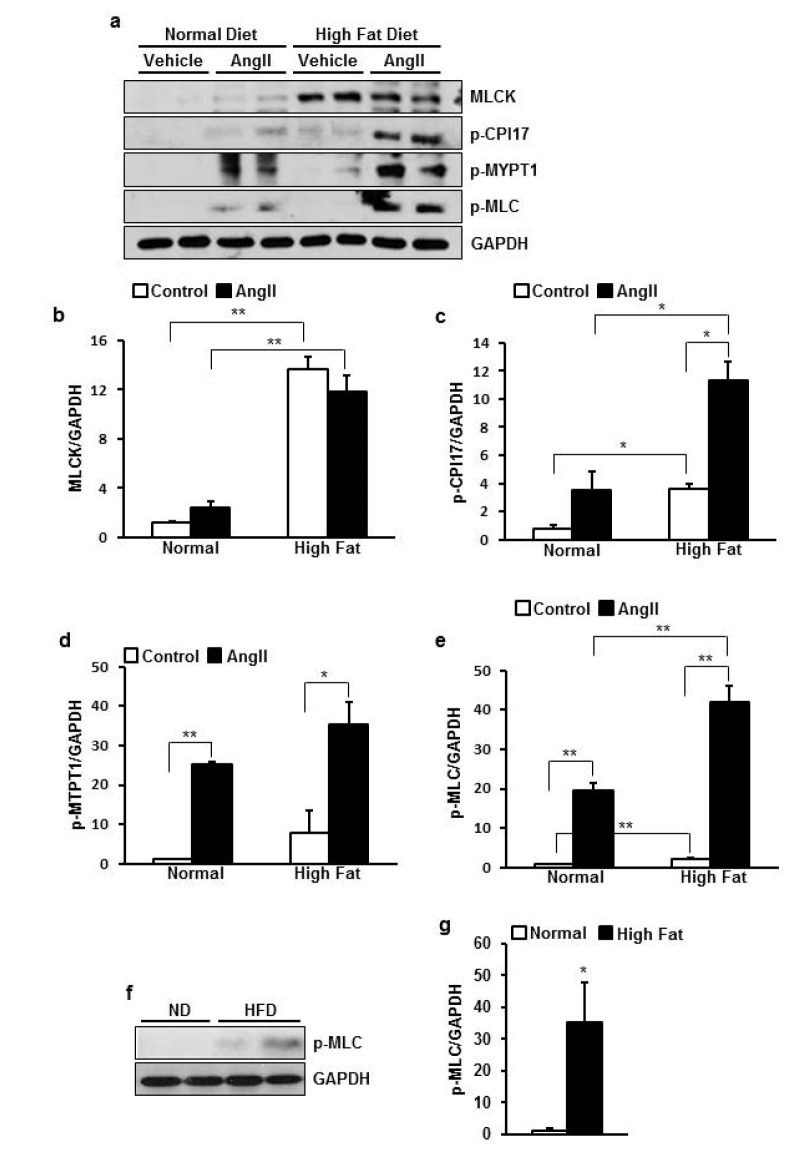
HFD increased basal phosphorylation of MLC via upregulation of MLCK and CPI-17 in rats
In order to investigate mechanism underlying increased vascular tone we examined expression and phosphorylation status of regulator proteins for the vascular contraction with/without HFD and Ang II stimulation. As a result, basal MLCK expression and p-CPI-17 were greatly higher in HFD group than ND group (p=0.003, 0.011 respectively) (Fig. 5b). Ang II-induced phosphorylation of CPI-17 were greatly higher in HFD group than ND group (p=0.025) (Fig. 5c). MYPT1 had a tendency of higher phosphorylation in HFD group than ND group (Fig. 5d). Consequently, MLC phosphorylation induced by Ang II was much greater in HFD group than ND group (p=0.020) (Fig. 5e) supporting enhanced contraction in HFD group.
Because basal MLCK and CPI-17 were upregulated, I checked that basal MLC phosphorylation was also increased in HFD group. I used 3 times more protein sample because the basal phosphorylation level of MLC was very low. Because of the difference in magnitude between basal and Ang II-stimulated phosphorylation, I retested only basal level of phosphorylation in ND and HFD group. As a result, basal MLC phosphorylation was 35 times higher in HFD group than ND group (p=0.020) (Fig. 5g, h).
DISCUSSION
Herein, I investigated the mechanism underlying HFD-induced hypertension and hyper-reactivity in response to Ang II. The most important new finding of this study is that HFD upregulated VSM contraction regulatory proteins such as MLCK and MLCP inhibitor protein CPI-17 in both resting and Ang II-stimulated condition resulting in increased Ang II-induced VSM contraction and BP. Our data suggest that MLCK and CPI-17 could be a novel target for the treatment of hypertension in obese individuals particularly induced by HFD, which is one of the most common causes of obesity in modern society.
Because rapidly growing obese population is highly associated with increase in serious cardiovascular disease and hypertension with consequent disability and early death [119], treating obesity-induced hypertension is very important. Based on small controlled trials and clinical experiences angiotensin converting enzyme inhibitor, thiazides, calcium channel blockers, and statins turned to be useful in treating obesity-related hypertension [20]. However, these drugs draw adverse effects including tolerability [21], renal dysfunction [22], angina, arrhythmias, hypotension, arthritis, asthma etc. [23]. Therefore, more specific targets are needed to treat obesity-induced hypertension safely and effectively.
It is well known that circulating RAAS components are increased and positively correlated with BP in the obese hypertensives [2425]. Our result also demonstrated increased plasma Ang II level in HFD-fed mice. It has been reported that Ang II involves in HFD-induced hypertension through various pathways including leptin-related nephropathy [26], restoration of sympathetic over-reactivity [27], megalin-dependent uptake of angiotensinogen into adipocytes [28], vascular oxidative stress and endothelial dysfunction [29], and increased VSM reactivity [131430]. Increased VSM reactivity to Ang II in HFD-fed individuals is reported to be attributed to increased expression of AT1R and ACE 2 [13], and reactive oxygen species (ROS) overproduction and enhanced release of Ca2+ induced by Ang II [14]. However drugs inhibiting most of these upstream mediators of Ang II have undesirable effects as mentioned here earlier. Thus in this study, VSM contractile proteins as downstream effectors of Ang II activation in HFD-induced hypertension was studied.
Activation of AT1 receptor by Ang II in VSM causes phospholipase C activation leading to generation of inositol triphosphate (IP3) and diacylglycerol (DAG), that activates intracellular Ca2+ release and protein kinase C respectively [31]. Increased Ca2+ activates MLCK resulting in MLC phosphorylation and VSMC contraction [1631]. In the present study, MLCK expression was increased by HFD, likely enhancing this event. I presume that it maintains vasoconstriction and blood pressure elevation. On the other hand PKC enhances potency of CPI-17 to inhibit MLCP by approximately 1000 folds through phosphorylating threonine 38 residue of CPI-17 [32]. The results also revealed that both basal and Ang II-induced phosphorylation of CPI-17 in HFD group was greatly higher than ND group explaining increased phosphorylated MLC and blood pressure.
The data showed that 16 weeks of HFD significantly increased plasma LDL cholesterol concentration while other lipids did not change suggesting moderate condition of metabolic syndrome. LDL cholesterol is known to cause impairment of vascular relaxation via superoxide-mediated endothelial dysfunction in metabolic disease such as diabetes [33] suggesting that LDL cholesterol also likely affected abnormal vascular tension in HFD-fed rats in this study.
In conclusion HFD increased plasma Ang II level, MLCK expression and inhibited MLCP by activation of CPI-17 resulting in enhanced MLC phosphorylation. I suggest that enhanced basal MLC phosphorylation and consequent VSM hyper-contractility by increased level of Ang II is responsible for HFD-induced hypertension.
 XML Download
XML Download