

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Lung cancer is a primary malignancy of the lungs and the leading cause of cancer-related death worldwide. In 2010, lung cancer had a mortality of 15 million, accounting for approximately 19% of all cancer-associated death [1]. There are two main types of lung cancers including the small-cell and non-small cell lung cancer (NSCLC), with the latter being the majority representing near 80% of all lung cancers. The current recommendation for managing early stage NSCLC is surgery (wedge resection, segmentectomy, or lobectomy depending on the extent of tumor involvement). This regimen can largely prevent cancer recurrence or metastasis and improve overall outcome. Unfortunately, due to the indolent course of NSCLC, many patients present with advanced stage tumors at initial diagnosis [2]. Chemotherapy with cisplatin, paclitaxel, doxorubicin and gemcitabine agents are used for these poor surgical candidates, and often combined to improve efficacy and reduce toxicity [3456]. However, systemic chemotherapy is associated with multiple detrimental effects such as bone marrow suppression, neurotoxicity, and hand-foot syndrome. Studies suggest it is largely attributed to the intravenous route of drug administration and poor organ selectivity of the therapeutic agents [78].
Docetaxel (DTX) is a second-generation semi-synthetic taxane that regulates actin dynamics during the cell division. DTX has significant anti-tumor activities against an array of human malignancies, and has been suggested as one of the most effective single agents for NSCLC [91011]. Docetaxel is mainly metabolized in the liver and excreted in the feces. A number of studies have suggested that CYP3A4 and CYP3A5, two members of the Cytochrome P450 3A subfamily, play crucial roles in the oxidation and elimination of DEX [12]. While the data lacks on CYP3A5, CYP3A4 has been known to be expressed in adult liver and intestine [13]. CYP3A4 expression shows a notable interpatient variability which may account for the differences in the clearance of DEX and DEX associated toxicity [14]. As with other non-selective agents, DTX has a number of serious side effects due to systemic use that had limited its clinical effectiveness. In the last decade, there has been a growing interest towards liposome-mediated drug delivery due to its advantages including biodegradability, biocompatibility, low toxicity, ease to control drug release, and enhanced organ targeting [151617]. Although appealing, there were still barriers for those formulations to meet the need for clinical use and industrial production. For example, large diameter (>7 µm) liposomes produced through conventional techniques were associated with thrombosis, a risk enhanced by plasma viscosity in patients with some types of cancers [18].
Against this background, we developed a patented DBaumNC technology by combining the solid dispersion and effervescent techniques. The produced liposomes are at approximately 1 µm in diameter and stable in different conditions [19]. Our earlier animal model studies suggested that DTX-LP has a high and rapid pulmonary-targeting effect, longer half-life of elimination from circulation, and less side effects compared to the injectable DTX (DTX-IN) [20]. Given that liposome carriers may alter biological behaviors of the drug, in this study we aimed to determine the metabolism and excretion of DTX-LP using the in vitro and in vivo animal experimental models.
METHODS
Reagents and animals
Docetaxel (DTX, >99.5%) was purchased from China National Biotec Group (Beijing, China). Docetaxel injection (DTX-IN) was purchased from Jiang Su Heng Rui Medicine Corporation (Lian Yun Gang, China). Docetaxel liposome (DTX-LP) was prepared according to our published method [19]. Paclitaxel (>99.0%) was acquired from Chongqing Meilian Pharmaceutical (Chongqing, China). NADPH and formic acid were purchased from Sigma-Aldrich (St Louis, MO, USA). Acetonitrile and methanol were purchased from Honeywell Burdick & Jackson (Mundelein, Illinois, USA). New Zealand rabbits (2.0±0.1 kg) were supplied by the animal facility of Chongqing Medical University under the Institutional Review Board (IRB) protocol SCXY 2007-0001.
Ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS)
The UPLC-MS/MS was performed on an Agilent 1290 Infinity UPLC (Agilent, CA, USA) coupled with an Agilent 6538 Q-TQF mass spectrometer. The chromatography to determine DTX metabolites is detailed as follows and in Supplementary Table 1: chromatographic column: Waters XSELECT HSS T3 (2.5 µm, 100×2.1 mm); mobile phases: phase A: 0.1% formic acid in water; phase B: 0.1% formic acid in acetonitrile; running condition: sample volume: 4 µL; flow rate: 0.4 mL/min; duration: 10 min; column temperature: 40℃. The mass spectrometry was run at the positive electrospray ionization (ESI+) mode with the parameters outlined in the Supplementary Table 2.
DTX in vitro metabolism
The in vitro experiments included three groups: liver homogenates, lung homogenates, and liver microsomes. Liver and lung were isolated from the New Zealand rabbits fasted overnight. The organs were rinsed with phosphate-buffered saline (PBS, 0.1 M, pH 7.4) containing 0.15 M KCl to remove contamination from the blood. The organs were homogenized with a Sonics VCX 130 ultrasonic processor (Newtown, CT, USA) in four volumes of PBS at 4℃. Total protein concentration was determined using the BCA assay according to the manufacturer's guidelines (Thermo Fisher Scientific, Springfield, NJ, USA).
1 mL of liver homogenates, lung homogenates or liver microsomes at a total protein of 1 mg/mL was transferred to the sterile tissue culture plates and mixed with DTX-IN or DTX-LP to a final drug concentration of 10 µg/mL. The reaction was allowed for 10 min at 37℃ water-bath under gentle oscillation. After adding 10 µL of 10 mM NADPH, the reaction was stopped at 0, 0.25, 0.5, 1, 1.5 or 2 hours by the addition of a triple volume of cold methanol containing 20 ng/mL paclitaxel. The mixture was vortexed and centrifuged at 4000 RPM for 10 min. 100 µL supernatant was taken and mixed with 300 µL purified water and centrifuged again at 13000 RPM for 5 min. DTX in the resulting supernatant was determined with the UPLC-MS/MS.
Determination of DTX metabolites
DTX-IN or DTX-LP stock solution was added to 1 mL of liver homogenates (1 mg/mL total proteins) to achieve a final diluted drug concentration of 10 µg/mL. Upon adding 10 µL of 10 mM NADPH, the homogenate was incubated at 37℃ water-bath under oscillation. The reaction was terminated at 0, 1.5 or 2 hours with two volumes of cold methanol. The solution was vortexed and centrifuged at 12000 RPM for 10 min. The supernatant was dry evaporated at 40℃ with a DCY-16S termovap nitrogen sample concentrator (Haike Instruments, Qingdao, China). The specimen was then reconstituted with methanol and centrifuged again at 13000 RPM at 4℃. The final supernatant was used for the UPLC-MS/MS analysis.
DTX in vivo metabolism
The in vivo experiments included two groups. Group A rabbits were injected with 1 mg/kg of DTX-LP via the auricular vein. Group B was injected with an equal amount of DTX-IN. Bile juice was collected at three time points including 10 mins prior to injection, 1 hour, or 2 hours after drug administration. Bile was centrifuged at 13000 RPM for 5 min, and the supernatant was subjected to the UPLC-MS/MS analysis.
DTX excretion study
1 mg/kg of DTX-LP or DTX-IN was administered to the A or B experimental group rabbits (5 each) by intravenous infusion. At the 4 h, 8 h, 16 h, 24 h or 48 h time point, animal urine and feces were collected. Urine was measured, centrifuged, and the supernatants were saved for further analysis. Feces was dried, weighed, and mixed with purified water at a ratio of 1:5 (g/mL). The specimens were mixed with 300 µL of ethanol containing 50 ng/mL of paclitaxel. The solutions were vortexed and centrifuged at 15000 RPM for 10 min. 320 µL of supernatant was taken and mixed with 80 µL of purified water. The final solution was mixed and submitted for the UPLC-MS/MS analysis.
DTX excretion and calculation of the cumulative excretion rate
The concentration of DTX in feces or urine was determined with the method as described above. Two equations were used to determine the DTX excretion. First, amount of DTX in feces or urine=DTX concentration in feces or urine×collected feces or urine volume. Second, cumulative excretion rate of DTX=cumulative amount of DTX in feces or urine within 48 hours/total dose of the drug given.
RESULTS
In vitro metabolism of DTX-LP
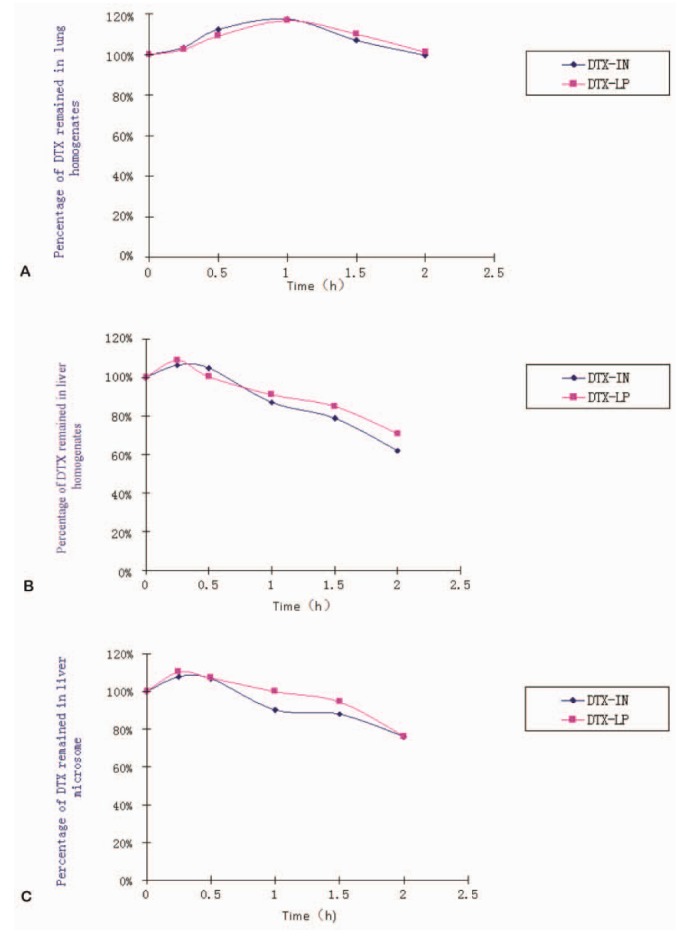
We first examined the in vitro metabolism of DTX-LP in liver homogenates, lung homogenates and liver microsomes using DTX-IN as the control. We calculated the peak area of DTX at various time points of incubation, and compared to the initial peak area. The metabolism profile of DTX-LP was quantified as the percentage of intact DTX at different time points (Fig. 1 and Table 1). With 2 hours of intubation, we noted no significant metabolism of DTX-LP or DTX-IN in the lung homogenates. By contrast, DTX-LP and DTX-IN were degraded by approximately 29.1% and 38.0% in the liver homogenates, and approximately 23.7% and 23.9% in the liver microsomes, respectively. Consistent with multiple reports, the decrease of DTX-LP and DTX-IN in the liver homogenates and microsomes indicated the metabolism of DTX by liver [1421]. DTX was not metabolized by the lungs; this could be associated with the lack or insufficiency of metabolic enzymes present in the lungs. Importantly, our data indicates that in vitro metabolism of DTX-LP appears similar to that of the intravenous agent DTX-IN.
In vitro metabolites of DTX-LP
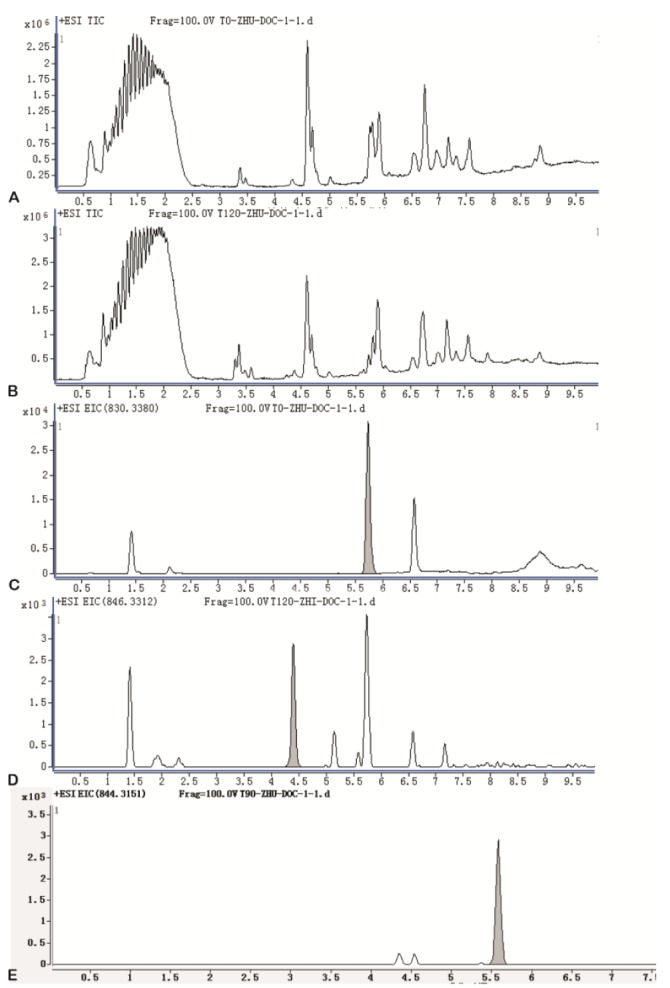
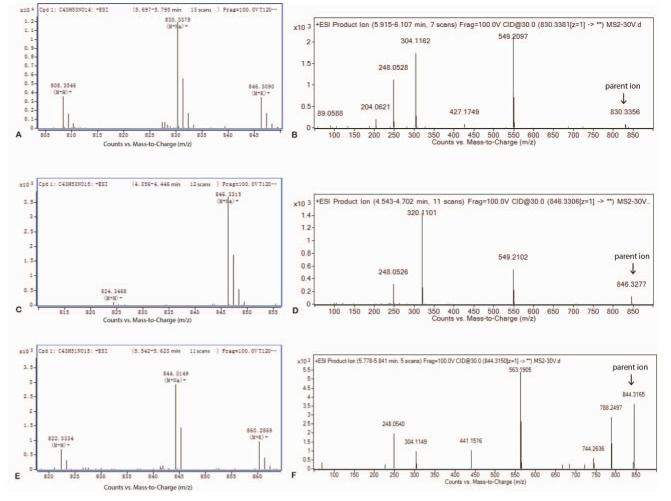
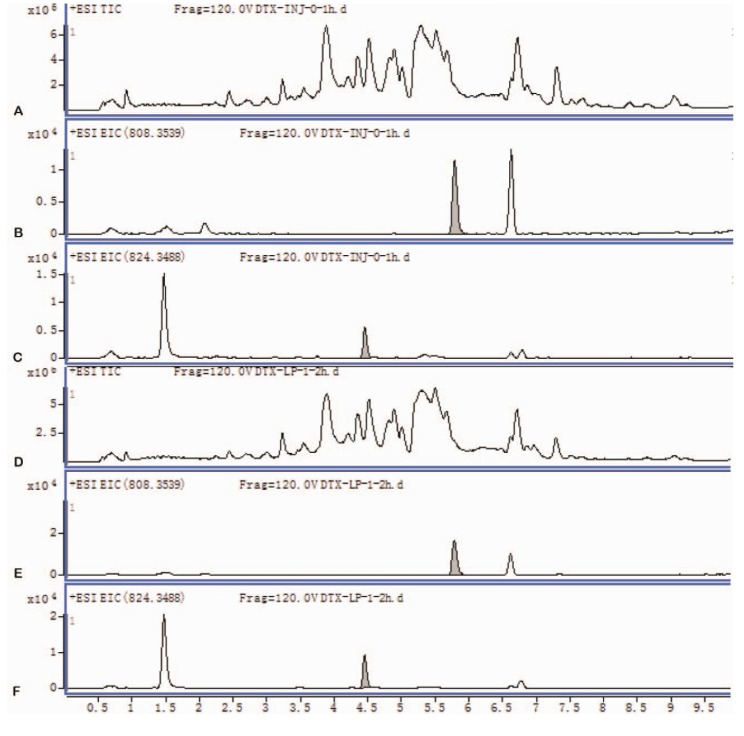
To determine the in vitro metabolites of DTX-LP, we incubated DTX-LP or DTX-IN with rabbit liver homogenates. After 2 hours of incubation, we detected two identical metabolites with UPLC-MS/MS (Fig. 2 for DTX-IN and Fig. 3 for DTX-LP). The first metabolite was the known M-2 with a molecular weight of 823. The second metabolite was a novel product which we named Mun (un for unconfirmed) with a molecular weight of 821. With further analyses, we found in the first order of mass spectrum, the massto-charge ratio (m/z) of adduct ions of DTX [M+H]+, [M+Na]+ and [M+K]+ was 808, 830 and 846, respectively (Fig. 4A). The secondary mass spectrometry detected the characteristic peaks of DTX fragments at 89, 204, 248, 304, 427 and 549, with the parent ion [M+Na]+ at 830 (Fig. 4B).
For metabolite M-2, the first order mass spectrum detected the [M+H]+ and [M+Na]+ at 824 and 846 (Fig. 4C). The secondary mass spectrometry detected the fragment peaks at 248, 320 and 549 with the parent iron [M+Na]+ at 846 (Fig. 4D).
For metabolite Mun , the [M+H]+, [M+Na]+ and [M+K]+ peaks were identified at 822, 844 and 860, respectively (Fig. 4E). In addition, the characteristic peaks of the Mun metabolites in the secondary mass spectrometry were present at 248, 304, 441, 563, 744 and 788, with the parent ion [M+Na]+ at 844 (Fig. 4F).
Finally, we compared the temporal metabolism of DTX-LP with DTX-IN at three different time points (0, 1.5 and 2 hours). Our results indicated that DTX, M-2 and Mun were present at the comparable levels for DTX-LP and DTX-IN (Table 2). At 1.5 hours, DTX-LP was present at 75.5±1.5% with the M-2 and Mun metabolites at 16.8±1.8% and 7.6±0.4%. At 2 hours, DTXLP remained at 72.6±2.3% with the M-2 and Mun metabolites at 18±2.8% and 9.5±1.4%. DTX-LP showed no significant difference when compared to the temporal metabolism of DTX-IN. At 1.5 hours, DTX-IN was maintained at 73.7±0.3%, and M-2 and Mun at 7.9±0.1% and 8.4±0.4%. At 2 hours, DTX-IN was present at 69±1.4% with M-2 and Mun at 19±0.5% and 12±1.8%, respectively. The subtle retention of DTX-LP in the rabbit liver homogenates likely reflects the process of DTX release from the liposomes.
In vivo metabolism of DTX-LP
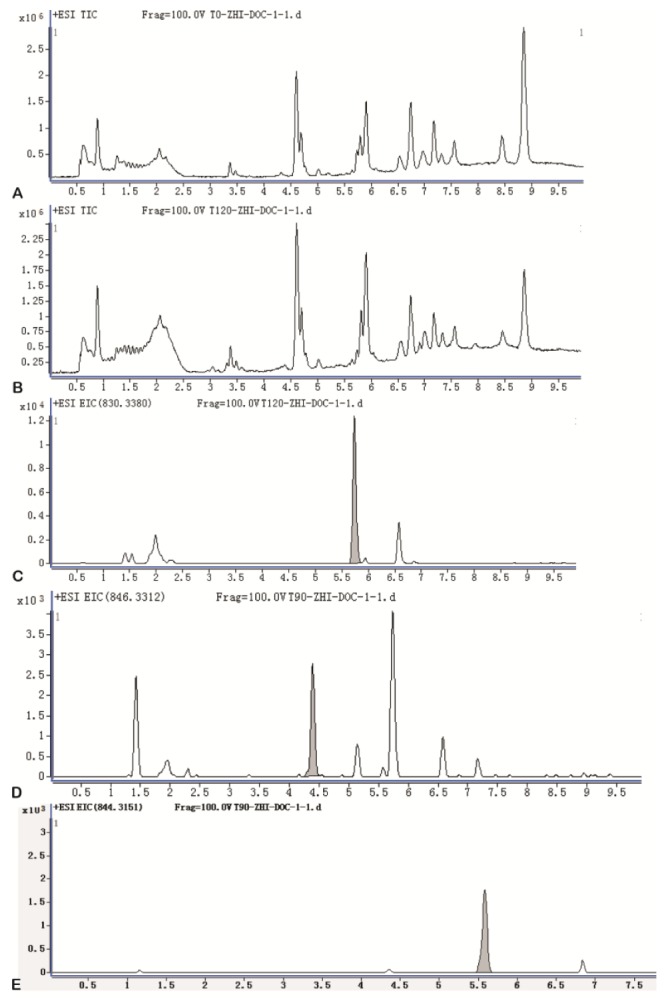
With the knowledge of in vitro metabolism, we next analyzed the in vivo metabolites of DTX-LP along with DTX-IN. We identified only one metabolite in the rabbit bile with a molecular weight of 821, consistent with M-2. The Mun metabolite was not identified at the 1 mg/kg given dose. However, with the increased dose at 2 mg/kg in a separate toxicokinetics analysis, we were able to detect the Mun in the rabbit feces (Results not shown). Fig. 5 illustrated the UPLC chromatograms of DTX-IN (Panel A~C) and DTX-LP (Panel D~F).
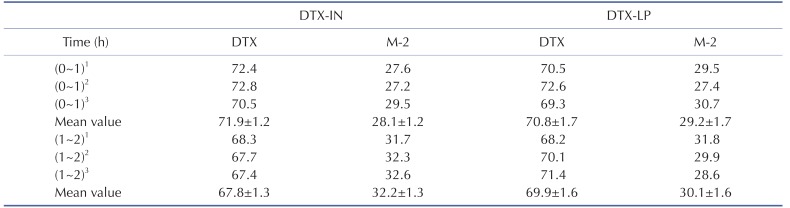
We further compared the proportion of DTX and M-2 metabolite at two time points (1 and 2 hours with 0 hour as the 100% baseline) (Table 3). We noted no significant difference in the in vivo metabolism of the intravenous and liposome forms of DTX. At 1 hour, DTX and M-2 in the DTX-IN group were present at 71.9±1.2% and 28.1±1.2%, while the DTX and M-2 in the DTX-LP group showed 70.8±1.7% and 29.2±1.7%. In addition, at 2 hours, DTX and M-2 in the DTX-IN group were 67.8±1.3% and 32.2±1.3%, and in the DTX-LP group were 69.9±1.6% and 30.1±1.6%.
Excretion of DTX-LP
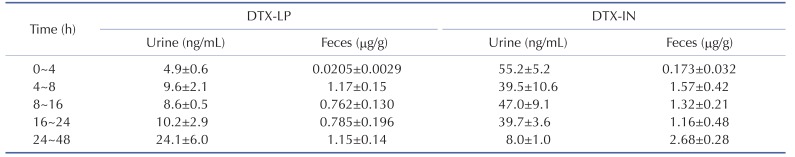
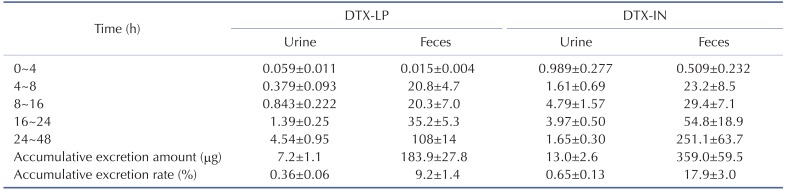
To determine the excretion of DTX-LP, we first compared the DTX present in rabbit feces or urine after intravenous injection of the two agents. DTX-LP showed significantly diminished excretion in feces or urine when compared to DTX-IN (Table 4). The amount of DTX-LP presented in urine was 4.9±0.6, 9.6±2.1, 8.6±0.5, 10.2±2.9 and 24.1±6.0 ng/ML at 4, 8, 16, 24 and 48 hours, respectively. In comparison, DTX-IN showed a much larger amount at 55.2±5.2, 39.5±10.6, 47.0±9, 39.7±3.6 and 8.0±1.0 ng/ML. Similar results were observed from the feces. The amount of DTX-LP detected was 0.0205±0.0029, 1.17±0.15, 0.762±0.130, 0.785±0.196 and 1.15±0.14 µg/g after 4, 8, 16, 24 or 48 hours. On the other hand, DTX-IN showed a larger excretion at 0.173±0.032, 1.57±0.42 µg/g, 1.32±0.21, 1.16±0.48 and 2.68±0.28 µg/g. We quantified the excretion of DTX by calculating the accumulative excretion amount and accumulative excretion rate with the methods detailed in the materials and methods. The accumulative excretion amount of DTX-IN at 48 hours was 359.0±59.5 µg in feces and 13.0±2.6 µg in urine. The cumulative excretion rate was 17.9±3.0% in feces and 0.65±0.13% in urine. In comparison, the accumulative excretion amount of DTX-LP was only 183.9±27.8 µg in feces and 7.2±1.1 µg in urine. Consistently, the cumulative excretion rate was only 9.2±1.4% in feces and 0.36±0.06% in urine (Table 5).
DISCUSSION
Non-small cell lung cancer (NSCLC) is the most common type of lung cancer and the leading cause of cancer-related deaths worldwide. With systemic chemotherapy, significant progress has been made to the clinical treatment of high stage NSCLC. However, chemotherapy-associated toxicity still presents a challenge to the oncologists. There is a continued demand for a targeted approach with improved therapeutic index and minimized systemic effects.
Docetaxel liposome (DTX-LP) is a newly-patented antimitotic chemotherapeutic agent. The negatively-charged DTX-LP engineered through the solid dispersion and effervescent techniques has been showed to have a favorable lung-targeting effect [1922]. The present study compared DTX-LP to the intravenous formulation DTX-IN to determine the metabolism and excretion in a rabbit experimental model. Our results suggest that DTX-LP and DTX-IN were metabolized in liver homogenates and microsomes, but not degraded by lung homogenates. We noted no significant difference in the in vitro or in vivo metabolism of DTX-LP and DTX-IN. However, liposomal delivery of DTX significantly delayed the drug excretion in rabbit urine and feces compared to the intravenous formulation. This would likely result in a prolonged accumulation of DTX in the lungs, facilitating the drug to fully exert its cytotoxic activity against NSCLC.
Interestingly, our triplicated experiments showed some “protective” effects of DTX by the lung homogenates (Table 1 and Fig. 1). For example, the DTX-IN after 1 hour of incubation was 117.6% and DTX-LP was 116.8%. These increases likely have represented the random errors produced by our small observational study with limited scientific and/or clinical relevance. Another possibility is certain factors (proteins, nucleic acids, etc.) from the lung homogenates might have interfered with our assay causing falsely increased concentrations. Lastly, the biotransformation/modification of the drug in lung homogenates could render the drug temporally resistant from the metabolic degradation. This, however, is a pure speculation. We performed a literature search and found one study with the similar findings [23]. Unfortunately, no specific explanation was given by the authors.
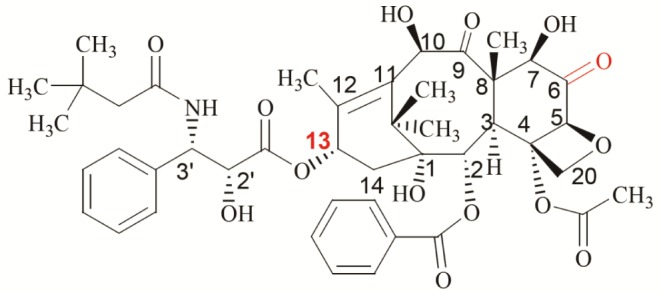
With ultra-performance liquid chromatography and tandem mass spectrometry, we identified two identical DTX metabolites (M-2 and Mun) from DTX-LP and DTX-IN. This finding suggests that there was no alteration in the metabolism of DTX-LP. To date, 10 DTX metabolites have been reported (See Supplementary Table 3). M-1, M-2, M-3 and M-4 are the major metabolites (Structures illustrated in Supplementary Fig. 1). In addition to the known M-2 metabolite, our mass spectrometry identified an unknown metabolite Mun with a molecular weight of 821. The structure of Mun differed from the reported M-1/M-3, peak 17 or peak 18 which had an identical molecular weight. M-1/M-3 is a pair of enantiomers generated from the dehydrocyclization of hydrogen atoms from the nitrogen atom of the M-2 C-13 side chain and the adjacent tert-butyl-hydroxymethyl carbon atom (Illustrated in Supplementary Fig. 2). The characteristic peaks of M-1/M-3 in secondary mass spectrometry were 296 and 527 ([m+H]+), indicating a break in the C-13 side chain resulting in a C-13 side chain (296) and a baccatin structure (527, [m+H]+) [24]. One of the characteristic peaks of Mun (563, [m+Na]+) was higher than M-1/M-3 (527, [m+H]+) by 14. One possibility of this difference in molecular weight was the addition of one oxygen atom and loss of two hydrogen atoms. With the obtained information, we proposed a most plausible structure of Mun in Fig. 6. It became visually evident that Mun differed structurally from M-1/M-3. Further, break of C-13 side chain of the peak 17 or 18 metabolite likewise would produce a characteristic peak of baccatin structure (527, [m+H]+). Given that this was not seen for Mun, it was unlikely that Mun shared the same chemical structure as peak 17 or 18.
As we know, the four major metabolites of DTX (M-1 to M-4) were all produced by the enzymatic action of cytochrome P450 CYP3A enzyme on the C-13 side chain [25]. The identification of Mun with a differing change to the baccatin structure has therefore suggested a possible alternative metabolic pathway. One possibility lies in the rabbit animal model we used for the study. Although studies have indicated a conservation of DTX metabolism in rats and humans, the mouse studies otherwise identified a few novel metabolites with changes to the baccatin structure [24]. These metabolites had the characteristic peaks 12 and 18 (525, [m+H]+), 2 fewer hydrogen atoms than the Mun (527, [m+H]+). Thus, they likely represented different DTX metabolites. Interestingly, paclitaxel, a same class taxane diterpenoids, has a significant species difference in metabolism. Paclitaxel is catalyzed to 6α-hydroxyl paclitaxel through the action of cytochrome CYP2C8 enzyme in humans. In opposition, species such as rats, rabbits, and pigs have the 3′-p-hydroxyl paclitaxel as an alternative product [26]. Thus, the true identity of Mun, whether it is specific for rabbits, and what enzyme is involved in the catalytic process remain interesting to be determined. It is within our future scope to purify Mun with liquid chromatography and determine its structure through methods such as nuclear magnetic resonance and infrared spectroscopic techniques. Further, with various inhibitors of cytochrome enzymes, we hope to pinpoint the enzyme responsible for the production of Mun
The low urinary or fecal excretion of DTX-IN in our study suggests that DTX-IN is not eliminated through the urinary system or gastrointestinal tract. This is consistent with multiples reports suggesting that DTX is mainly excreted in the forms of metabolites [2526]. However, the fecal excretion rate of DTX-LP at 9.2% or DTX-IN at 17.9% was still higher than the literature. We speculate this could again be caused by the species difference, wherein most other studies used mice or dogs as opposed to our rabbits [1225272829]. The cumulative excretion rate of DTX-LP was significantly lower than DTX-IN. This was anticipated given that DTX-LP was more concentrated in the lungs delaying the drug elimination.
While the experimental animals do not completely recapitulate the human physiology, our New Zealand rabbits is a well-established preclinical research model with good human correlation. New Zealand rabbits have a few advantages for the translational research given they are non-aggressive and economical animals with a relative short vital cycle. Rabbit studies provide vital preliminary information such as drug distribution, clearance, bioavailability and excretion in the form of metabolites. With that information, the phase I clinical trials ultimately determine the drug safety, dosage and common side effects in a small cohort of patients.
Collectively, our study found that liposomal delivery of DTX did not alter the in vitro and in vivo drug metabolism. This ensures its pharmaceutical safety for clinical use. The identification of Mun suggests a possible alternative metabolic pathway in the rabbits. We also confirmed that pulmonary-targeting DTX-LP was not degraded by the lungs and had delayed excretion compared to DTX-IN. This will greatly enhance the DTX therapeutic efficacy and reduce the systemic side effects for the treatment of NSCLC. As initial evidence, our ongoing study with the preclinical VX2 rabbit lung cancer animal model has found that 50% of DTX-LP was sufficient to double the animal survival with significantly reduced side effects.
 XML Download
XML Download