

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Calcium channel blockers (CCBs) were developed in the 1970s and are now widely used for cardiovascular diseases such as hypertension and ischemic heart disease [1234]. Since CCBs also potently inhibit Ca2+ influx in arterial myocytes, they induce vascular relaxation and lowering of the blood pressure. CCBs are divided into several subtypes based on their chemical structures and functional mechanisms: the dihydropyridine (DHP), phenylalkylamine and benzothiazepine classes. According to clinical guidelines, the DHP CCBs belong to the recommended first-line antihypertensive drugs to treat essential hypertension [5]. Nicardipine (NIC, first-generation), isradipine (ISR, second-generation), and amlodipine (AML, third-generation) belong to the DHP-derivative group of CCBs (Fig. 1).
On the cardiac action potential (AP), voltage-gated Na+ channel current (INa) causes initial depolarization of upstroke phase, and thus open the voltage gated Ca2+ channel (VOCC) to open, allowing Ca2+ into the cell to prolong the AP and onto the sarcoplasmic reticular membrane to stimulate contraction through Ca2+-induced Ca2+ release (CICR) [6]. CCBs hinder calcium entry to the cardiac myocytes, thereby reducing the amount of Ca2+ available to induce CICR [7].
A unique feature of cardiac AP is the plateau phase of sustained depolarization that is due to both delayed activation of voltage-gated K+ channels currents (IKv) and VOCC current (ICa). Since the balance between ICa and IKv determines the amplitude and duration of the plateau phase of cardiac AP, pharmacological inhibition of the associated ion channels has been a critical issue of cardiac toxicity in terms of electrophysiology. IKv is composed of rapidly-activating and slowly-activating voltage-gated K+ channel currents called IKr and IKs, respectively. The inhibition of hERG (human ether-a-go-go-related gene) K+ channel, the major component of IKr is the most intensively investigated target [8]. Along with the decreased hERG activity due to either pharmacological agents or genetic mutations, suppression of slowly-activating voltage-gated K+ channel current (IKs) also induce the prolongation of action potential duration (APD) [9]. An abnormal APD prolongation predispose to arrhythmia due to early after-depolarization (EAD). In the heart, drug-induced QT interval prolongation in electrocardiogram (EKG) is recognized as potential risks such as torsades de pointes [1011]. Conversely, however, less is known about short QT syndrome. Nevertheless, genetic disorders or pharmacological side effects may induce abnormally short QT intervals that could potentially increase the risk of sudden death with atrial fibrillation and/or ventricular fibrillation [121314151617].
In contrast to the inhibition of K+ channels, decreased ICa is expected to shorten APD and/or modify the shape of plateau in the cardiac AP. Therefore, CCBs can theoretically cause APD shortening. However, the above CCBs are widely used without severe side effects. Such practical safety might be due to the low plasma concentrations of CCBs in the patients prescribed with CCBs against hypertension. Another possibility is a putative compensatory inhibition of K+ channels such as hERG, which might counterbalance the APD shortening effect of CCBs. However, precise investigations on the latter possibility is lacking yet. The purpose of this study was to examine the effects of NIC, ISR, and AML on the AP in rabbit Purkinje fibers and on cardiac ion channel currents, especially K+ channels currents associated with the repolarization process. The integrated analysis of cardiac ion channels might provide a novel insight to understand the pharmacological effects of CCBs without critical side effects in the clinical applications.
METHODS
Animals
The experiments for AP recording and ICa analysis were performed using New Zealand white rabbit (2.5~3.5 kg) and male Sprague-Dawley (SD) rats (250~350 g), respectively. The animals were kept in a storage room under the conditions of constant temperature (23±3℃), relative humidity (50±10%), and illumination (12 h light/dark cycles) until the initiation of the experiment. This study was conducted in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. All procedures were approved by our Institutional Animal Care and Use Committee (IACUC).
Cell preparation
To assess the effects of CCBs on IKr, IKs, IK1, and INa, HEK293 cells were transiently transfected with the following genes using Lipofectamin2000 (Gibco BRL, USA) according to the manufacturer's instructions. The hERG (the gene corresponding to IKr), KCNQ1/KCNE1 (the gene corresponding to IKs), KCNJ2 (the gene corresponding to IK1) or SCN5A (the gene corresponding to INa) cDNA was co-transfected with green fluorescence protein (GFP) to allow assessment of the transfection efficiency. The overexpression system was adopted for the analysis of pharmacological drug effects on the above ionic currents because reliable functional isolation of respective ion channel current in cardiomyocytes is technically difficult. Although the HEK293 cells endogenously express voltage-gated K+ currents, the peak amplitudes were less than 10% of the overexpressed IKr and IKs amplitudes.
To assess the effects of CCBs on the calcium currents, however, enzymatically isolated naïve ventricular myocytes were used because consistent co-expression of multilple subunits of L-type Ca2+ channel proteins were requested. Briefly, the hearts were rapidly excised from anesthetized SD rats and perfused via the aorta on a Langendorff apparatus with an oxygenated normal Tyrode's (NT) solution for 5 min to clear the blood, then perfused with Ca2+-free NT solution for 3 min. Next the heart was perfused with enzyme solution containing 0.6 mg/ml collagenase (Worthington, type 2, USA) for 30~40 min. Finally, this enzyme-containing solution was washed out for 5 min with a high-K+ and low-Cl- Kraft-Bruhe (KB) solution. Following the isolation procedure, the left ventricle was dissected out and agitated mechanically with a fire-polished Pasteur pipette in KB solution to obtain single myocytes. The isolated myocytes were stored at 4℃ until use for up to 8 hour.
Drugs and Solutions
NIC, ISR and AML were purchased on Sigma-aldrich (MO, USA). These were formulated into stock solution with dimethyl sulfoxide (DMSO). All the drug stock solutions were diluted in normal Tyrode (NT) solution to produce the target exposure concentrations. The concentration of DMSO in NT was always kept 0.1%. The external solution for recording the IKr, IKs and INa was NT solution as follows (in mM): 143 NaCl, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 5 HEPES, 0.33 NaH2PO4 and 16.6 glucose (pH adjusted to 7.4 with NaOH).
The internal solution for recording IKr contained the following (in mM): 130 KCl, 5 Ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid (EGTA), 10 4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES), 1 MgCl2, and 5 Mg-ATP (pH adjusted 7.25 with KOH). For recording IKs the internal solution contained (in mM) 150 KCl, 5 EGTA, 10 HEPES, 2 MgCl2, 1 CaCl2 and 5 Na2-ATP (pH adjusted 7.25 with KOH). For recording IK1, the internal solution contained (in mM) : 130 K-Asp, 15 KCl, 10 HEPES, 1 MgCl2, 5 Na2-ATP, 5 EGTA (pH adjusted 7.25 with KOH). For recording INa, the internal solution contained 105 CsF, 35 NaCl, 10 EGTA, 10 HEPES (pH adjusted to 7.25 with NaOH).
For recording ICa, the fresh isolated rat ventricular myocytes were superperfused with an external solution that consisted of (in mM): 137 cholin-Cl, 5 CsCl, 0.5 MgCl2, 2 4-AP, 10 HEPES, 10 glucose and 1.8 CaCl2 (pH adjusted to 7.4 with NaOH). The internal solution for ICa recording contained (in mM): 20 CsCl, 100 Cs-aspartate, 10 EGTA, 10 HEPES, 20 Tetraethylammonium chloride (TEA-Cl), 5 Mg-ATP (pH adjusted to 7.25 with KOH). KB solution for storage of the freshly isolated rat ventricular myocytes contained (in mM): 70 K-glutamate, 55 KCl, 10 HEPES, 3 MgCl2, 20 taurine, 20 KH2PO4, 0.5 EGTA (adjusted to pH 7.2 with KOH).
Recording of action potentials on rabbit Purkinje fibers
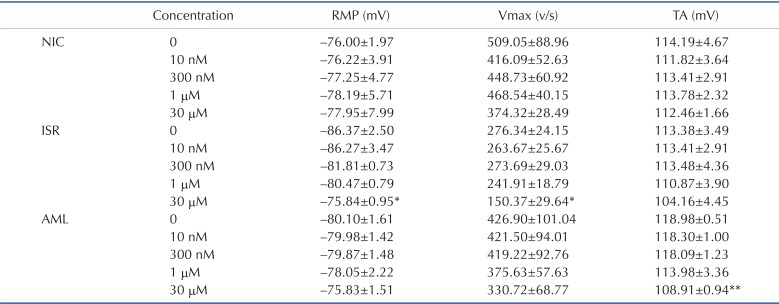
The rabbits were anesthetized with pentobarbitone sodium (30~50 mg/kg i.v.) and then their hearts were rapidly removed and placed in oxygenated NT solution to pump the remaining blood out. The left ventricle was opened and Purkinje fibers were carefully dissected out with a small piece of ventricular tissue to be pinned in the experimental chamber. The isolated Purkinje fibers were superfused with oxygenated NT solution (5 ml/min) maintained at 37.0±0.5℃. The preparations were electrically stimulated at a basal rate (frequency=1 Hz, duration=2 ms, voltage=1.5~2 V). Two hours were allowed for each preparation to equilibrate while continuously superfused with NT solution. Action potentials were recorded using the conventional intracellular recording technique involving a glass microelectrode filled with 3 M KCl and connected to a Geneclamp 500B (Axon Instruments, CA, USA). Action potential duration at 50% and 90% repolarization (APD50 and APD90) was automatically measured using Notocord HEM program (NOTOCORD, France) at a sampling rate of 50 kHz. Before drug treatment, action potential parameters were measured in NT for 1 hour to establish stable control value recording. The vehicle control (0.01% DMSO in NT) and drugs were perfused every 20 min after the stable AP were obtained. Besides AP, resting membrane potential (RMP), total amplitude (TA), and maximum velocity of initial depolarization (Vmax) were analyzed (Table 1). The RMP values in the Purkinje fibers usually lie between -89 and -72 mV. The tested fibers were randomly allocated to each test agent. However, since the numbers of tested tissues are not large, accidental gathering of relatively hyperpolarized Purkinje fibers for a specific test group could results in an outlier-like data (see the RMP for the ISR group in Table 1).
Recording of ionic currents
The cells were placed in a recording chamber on the stage of a Nikon inverted microscope, and continuously perfused (5±1 ml/min) with 37±1℃ bath solution. Ionic currents were recorded in a whole-cell configuration with a standard patch clamp technique using a HEKA EPC8 amplifier (Electronik, Lambrecht, Germany). Data were recorded during the approximately 5 minutes following initial application of the bath solution to verify currents stability. Test drug solutions were subsequently superfused for approximately 5 minutes to achieve steady-state blocks. To investigate the effect of these drugs on the ion channel currents, various concentrations (0.01~30 µM) of drugs were tested. Voltage-clamp protocol generation and data acquisition were controlled by computers equipped with an A/D converter, Digidata (Axon Inc., USA) and RClamp software developed by Seoul National University (Seoul, Korea). The patch pipettes were made from borosilicate glass capillaries (Clark Electromedical Instruments, UK) using a pipette puller (PP-830, Narishige, Japan). Their resistances were 3~4 MΩ when filled with pipette solution. The current signals were filtered at a sampling rate of 5 kHz, and they were low-pass filtered at 1 kHz and stored on computer. All experimental parameters, such as pulse generation and data acquisition, were controlled using the RClamp software.
Statistical Methods
Data analysis and curve fitting of patch clamp experiments were carried out using RClamp, GraphPad InStat (GraphPad Software, San Diego, CA), and SigmaPlot 2000 (SPSS Inc., Chicago, IL). Pooled data are expressed as means±standard errors of the mean (SEM), and statistical comparisons were made with p<0.05, or p<0.01 considered significant. Current amplitudes were measured before and after application of the respective drugs. The percent inhibition values were calculated according to the following equation:

Effects were calculated from the results of 3~5 experiments per concentration of the drugs. Concentration response relations were calculated by a non-linear least squares fit equation [Hill equation; f=xH/(IC50H+xH); H=Hill coefficient, IC50=IC50, x=concentration, f=inhibition ratio] using the SigmaPlot 2000 program for the half-maximum inhibiting concentration (IC50).
RESULTS
Effects of CCBs on ICa and INa
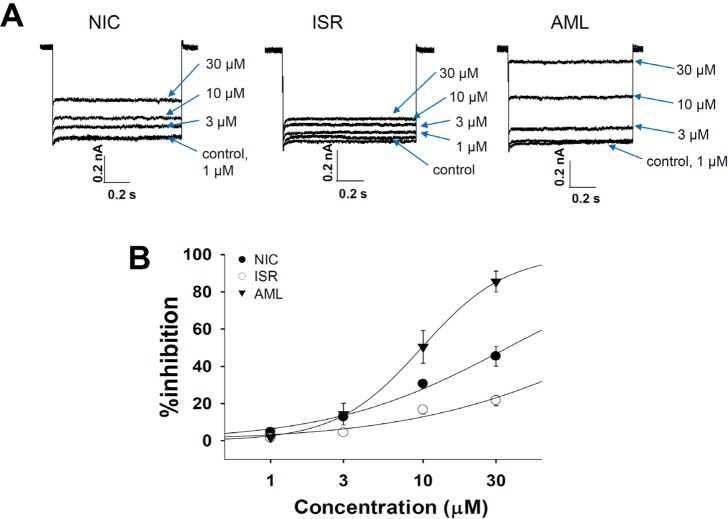
We first examined their inhibitory potency for Ca2+ channel currents in rat ventricular myocytes. The ICa was activated by a depolarizing step pulse (0 mV, 500 ms) from the holding potential of -80 mV (Fig. 2A). All three CCBs inhibited the ICa in a concentration-dependent manner (Fig. 2A). NIC at 0.01, 0.1, 1, and 10 µM reduced the ICa amplitude by 18.4%, 41.5%, 78.3%, and 99.4%, respectively (n=3). ISR at the same concentrations attenuated the ICa amplitude by -3.2%, 22.6%, 89.9%, and 99.8%, respectively (n=3). In addition, AML also had potent inhibitory effect on ICa. AML reduced the ICa amplitude by 13.8%, 34%, 72.5%, and 100% at 0.01, 0.1, 1, and 10 µM, respectively (n=3). The IC50 values were 0.142±0.03 µM for NIC, 0.227±0.28 µM for ISR, and 0.229±0.02 µM for AML (Fig. 2B).
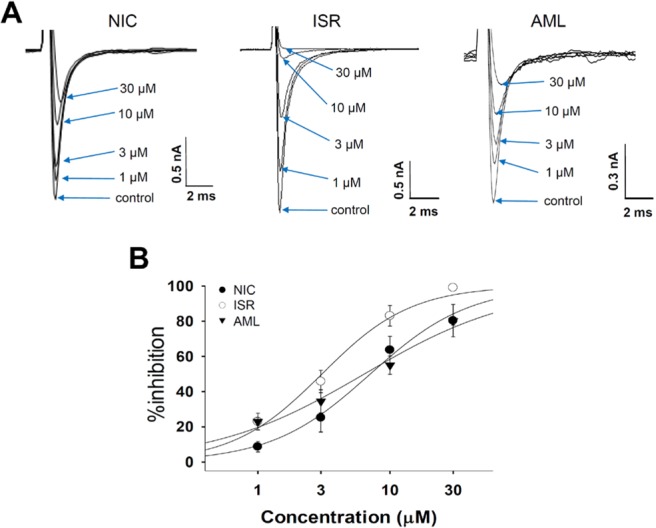
In SCN5A overexpressed cells, INa was generated by a step pulse from -100 mV of holding voltage to -40 mV of 20 ms duration. The CCBs also inhibited INa at micromolar ranges (Fig. 3A). 30 µM ISR almost completely inhibited INa while NIC and AML inhibited INa by about 80% at 30 µM. When fitted to a Hill function, their IC50 values were 7.08, 3.05, and 6.38 µM for NIC, ISR, AML, respectively (n=4, Fig. 3B).
Effects of CCBs on the cardiac APs in rabbit Purkinje fibers
ICH S7B guideline S7B recommends an integrated approach for cardiovascular preclinical evaluation of new drug candidates, including action potential assays with isolated cardiac tissues from guinea pig, rabbit, and canine to predict the potential of drugs to induce abnormal cardiac events. Among them, the AP assay using rabbit Purkinje fibers was considered a valuable assay for evaluating the proarrhythmic pharmaceuticals as it can reveal complex electrophysiological profiles that modulate repolarization delay [18]. To evaluate the integrated electrophysiological effects of CCBs, we performed rabbit Purkinje fiber assay with these CCBs.
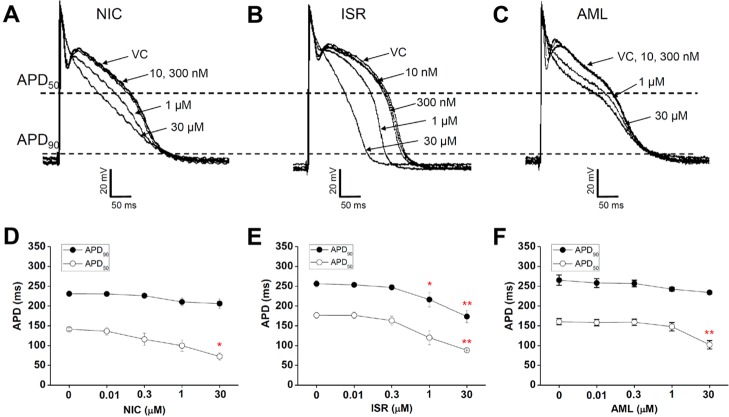
Fig. 4 shows the concentration-dependent effects of NIC, ISR and AML on the APs in rabbit Purkinje fibers. NIC at a concentration of 30 µM induced a triangulated AP and significantly shortened the APD50 compared vehicle control by -69 ms±9.9 while not affecting APD90 (n=4, p<0.05, Fig. 3A and 3D). The other AP parameters, including resting membrane potential (RMP), maximum velocity of initial depolarization in the phase 0 (Vmax) and the total amplitude (TA) were not changed (Table 1). AML at 30 µM also significantly shortened the APD50 by -58.3 ms±9.3 (n=4, p<0.01, Fig. 4C and 4F) while not significantly decreased the APD90. AML also decreased TA by -10 mV±0.93 (Table 1). NIC and AML induced a triangulated shape of AP (Fig. 4A and 4C). Unlike NIC and AML, ISR at 30 µM significantly shortened both the APD50 and APD90 by -88.5 ms±7.3 and -82.9 ms±8.8, respectively (n=4, p<0.01). ISR at 1 µM also markedly shortened the APD90 by -40 ms±17.8 (p<0.05, n=4, Fig. 4B and 4E). In addition, ISR at 30 µM significantly decreased the RMP (-10.5 mV±3.4, p<0.05) and Vmax (-126 V/s±10, p<0.05), but had no significant effect on the TA (Table 1).
Effects of CCBs on repolarizing K+ currents
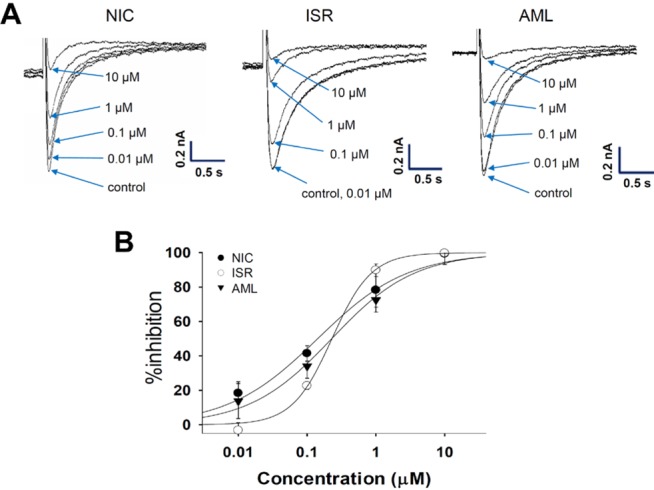
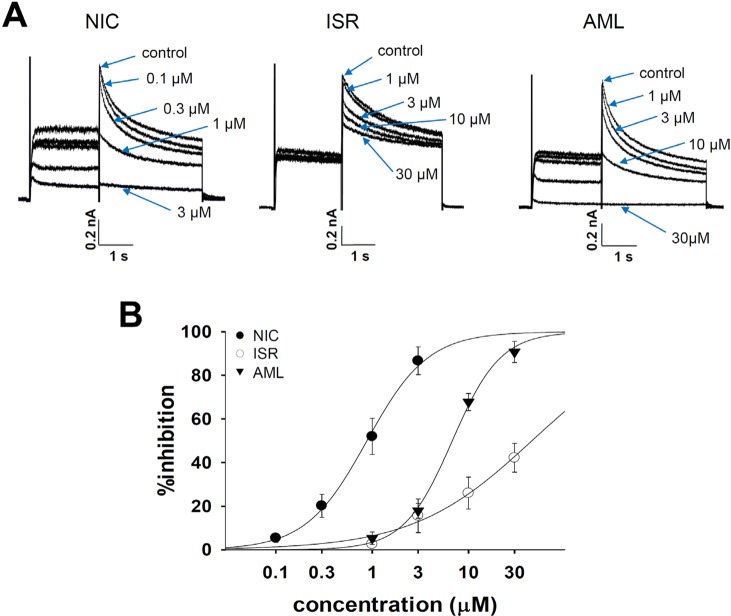
To examine the tail components of IKr which reflect the repolarizing K+ current in the cardiac AP, the cells were depolarized for 2 s to +20 mV from a holding potential of -80 mV followed by a 3s repolarization back to -40 mV. Fig. 4A shows the representative cases of the voltage-clamp recording from hERG-transfected HEK293 cells. NIC and AML commonly inhibited IKr in a concentration-dependent manner, and almost complete inhibition was observed at 30 µM (Fig. 5A and 5B). NIC at concentrations of 0.1, 0.3, 1, and 3 µM reduced the IKr amplitude by 5.1±2.9%, 13.0±2.5%, 40.1±5.5%, and 83.0±7.3%, respectively (n=4). AML at the same concentrations inhibited the IKr amplitude by 5.2±2.9%, 17.5±3.3%, 67.6±4.0%, and 90.4±5.1%, respectively (n=4). However, ISR at 1, 3, 10, and 30 µM inhibited the IKr amplitude by 2.6±1.6%, 15.6±7.7%, 26.0±7.3%, and 42.2±6.6%, respectively (n=4). The Hill equation fitting function was applied and IC50 values were measured to examine the relative potency of IKr inhibition (Fig. 5B). The IC50 values were 0.88±0.05 µM for NIC and 6.78±0.36 µM for AML. Since the maximum inhibition rate of ISR was smaller than 50%, we could not obtain the IC50 value for ISR.
For recording IKs, the KCNQ1/KCNE1-coexpressing cells were depolarized for 3 s to +60 mV from a holding potential of -80 mV, followed by a 3s repolarization back to -40 mV. Fig. 6A shows the representative current traces under control conditions and after exposure to 1, 3, 10, and 30 µM NIC, ISR, and AML. Similar to the effects on IKr, NIC and AML inhibited the IKs in a concentration-dependent manner while ISR incompletely inhibited IKs even at 30 µM. Fig. 6B shows the concentrationresponse curves for these drugs. The IC50 values were approximately 9.61±1.01 µM for NIC and 5.81±0.5 µM for AML. Since the maximum inhibition rate of ISR was smaller than 50%, we could not obtain the IC50 value for ISR.
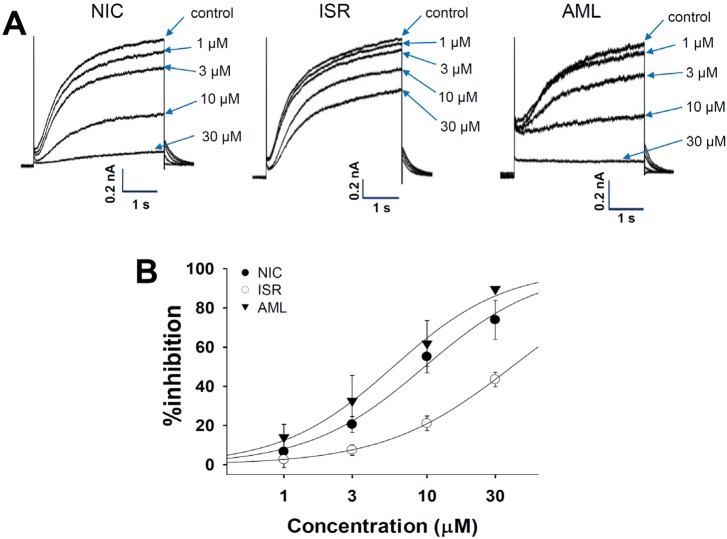
Fig. 7A shows the represented currents traces from KCNJ2-transfected HEK293 cells and the effects of CCBs. The IK1 was elicited by a hyperpolarizing step pulse from -80 mV to -120 mV of 1 s duration. All of the three drugs inhibited the IK1 in a concentration-dependent manner, however, the maximum inhibition rates of NIC and ISR at 30 µM were below 50% (45.4±5.3%, and 21.7±2.8%, respectively, each n=4). The IC50 value of AML on the IK1 was 9.78±0.18 µM (Fig. 7B, n=4).
DISCUSSION
Here we evaluated the electrophysiological safety of the most commonly used DHP class of CCBs by assessing their effects on the ion channel currents involved in cardiac APD and on repolarization phase in rabbit Purkinje fibers. Despite the common inhibitory effects on ICa, the APD shortening was not consistent between the three CCBs tested here. APD90 was significantly decreased only by ISR from 1 µM whereas APD50 was commonly decreased by NIC, ISR and AML at 30 µM. As a result, the AP was generally shortened by ISR whereas showed triangulation in response to NIC and AML. In addition to the above findings, ISR at 30 µM decreased the RMP and Vmax while not the TA (Table 1). The decrease of Vmax might be due to the more potent inhibition of INa by ISR than by NIC and AML (Fig. 3).
Except ISR, relatively high concentrations of NIC and AML were required to significantly decrease the APD90 of rabbit Purkinje fibers. Indeed, the present ICH S7B guideline [19] does not specifically address the possibility of a drug-induced shortening of the QT interval. Although the QT-shortening could potentially increase the ventricular tachycardia and the ventricular fibrillation risk of sudden death [2021222324], Roden [25] and Hondeghem [2627] have suggested that simple QT interval change is a poor marker for proarrythmic susceptibility. However, many experiments in isolated rabbit hearts demonstrated that triangulation (prolongation of the fast repolarization phase) is proarrhythmic [26] that was confirmed by other groups [28293031]. Triangulation may be accompanied by either shorting or lengthening of the total action potential duration. In this study, NIC and AML induced the triangulation of AP. However, relatively high concentration (e.g. 30 µM) was required to reveal the triangulation of AP, suggesting the safety of CCBs.
Several large clinical trials that have consistently shown that no significant increase in sudden cardiac death with DHP class of CCBs even in vulnerable patients [32333435]. In fact, the reported plasma concentrations of NIC, ISR and AML ranges below micromolar concentrations [363738] where no significant changes in APD were observed with NIC and AML in the present study. The DHP calcium channel modulators do not exert significant electrophysiologic effects on the myocardium and specialized conduction tissue at clinically relevant doses that dilate vascular smooth muscle in vivo. Owing to the relative selectivity of CCBs on vascular smooth muscle, significant depressant effects on myocardial contractility and atrioventricular conduction could be avoided [39]. However, the tissue-dependent selectivity can be lost in overdose situations [40]. In this respect, the potent inhibition of ICa without significant APD shortening by NIC and AML observed in the present study might imply an additional safety mechanism besides the plasma concentrations.
Here we suggest that the lack of actual clinical problems might be due, at least partly, to the concomitant changes in other ion channel activities. In addition to the inhibition of ICa, NIC and AML showed concentration-dependent inhibition of IKr and IKs; (Fig. 5 and 6). The inhibitory effects of ISR on IKr and IKs were incomplete (<50% at 30 µM). Since the voltage-gated K+ channels would mainly contribute to cardiac repolarization, their inhibition by NIC and AML might compensate for the putative APD shortening effects of these CCBs.
In summary, despite the potent ICa inhibition, NIC and AML do not induce a significant shortening of APD up to 30 µM of applied concentration. According to ion channel studies, concomitant inhibition of IKr and IKs might differentially counterbalance the influence to cardiac repolarization of CCBs. Further investigation of other classes of CCBs' effects on the cardiac K+ channel currents are requested for the integrative understanding of cardiac toxicity. Such study could include the physiological components having multiple effects on cardiac functions including ion channels [41]. Finally, our present study gives a message that the integrative analyses of various ion channel currents could provide a novel insight for the pharmacological effect as well as the safety for the clinical applications of drug compounds under development [42].
 XML Download
XML Download