

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Healthy vascular endothelial cells have distinct and unique regulatory functions affecting vascular tone, blood clotting, angiogenesis, and vascular inflammation [1]. However, if endothelial cells lose their function (endothelial dysfunction), atherosclerosis occurs. Vascular endothelial cells begin to express various adhesion molecules on their surface, including vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecules-1 (ICAM-1), and E-selectin, which play important roles in blood monocyte recruitment to the arterial intima during the early stage of atherosclerosis [2]. Reactive oxygen species (ROS) are a major risk factor for endothelial dysfunction because they control a number of signaling pathways relevant to atherosclerosis.
P66shc, a protein that belongs to the shcA family of adaptor proteins, has three isoforms with molecular masses of 46, 52, and 66 kDa (p46shc, p52shc, and p66shc, respectively). P66shc plays a key role in endothelial oxidative stress leading to endothelial dysfunction and is associated with pathophysiological conditions such as atherosclerosis. Cells lacking p66shc are resistant to apoptosis induced by H2O2, and mice deficient in p66shc show increased resistance to ROS and a 30% increase in life span [3,4]. The deletion of p66shc protects against age-associated endothelial dysfunction [4] and prevents hyperglycemia-induced endothelial dysfunction [5]. In addition, p66shc stimulates atherogenesis. Mice deficient in p66shc and fed a high-fat diet show reduced vascular cell apoptosis and early atherogenesis [6]. Similarly, p66shc mediates homocysteine-stimulated ICAM-1 expression and monocyte adhesion [7], as well as low-density lipoprotein-induced ICAM-1 expression and monocyte adhesion to endothelial cells [8].
Nafamostat mesilate (NM), a serine protease inhibitor, is used in patients with disseminative blood vessel coagulation, hemorrhagic lesions, and hemorrhagic tendencies. NM improves acute pancreatitis and prevents blood clot formation during extracorporeal circulation [9,10]. It also has an anti-inflammatory effect. Further, it inhibits lipopolysaccharide-induced nitric oxide production, apoptosis, and interleukin (IL)-6 and IL-8 levels in cultured human trophoblasts [11]. NM also prevents nuclear factor kappa-B (NF-κB) activation, induces caspase-8-mediated apoptosis in pancreatic cancer cells [12], and inhibits ICAM-1, vascular endothelial growth factor, and matrix metalloproteinase-9 activity as well as cell adhesion and invasion by inhibiting NF-κB during the peritoneal dissemination of pancreatic cancer [13]. However, the anti-inflammatory effects of NM on vascular endothelial cells have not been elucidated fully. Therefore, in the present study, we investigated the protective effects of NM on inflammation in vascular endothelial cells.
METHODS
Cell culture and cell viability assay
Human umbilical vein endothelial cells (HUVECs) were purchased from Clonetics (San Diego, CA, USA) and cultured in endothelial growth medium (EGM-2). Sub-confluent, proliferating HUVECs at passages 2~8 were used. U937 cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics. The effects of NM on HUVEC viability were examined using an ADAM-MC automatic cell counter (Digital Bio, Seoul, South Korea), which assesses propidium iodide (PI) staining [14].
Antibodies and immunoblotting
Anti-VCAM-1, anti-ICAM-1, and anti-β-actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-shcA antibodies were purchased from Becton Dickinson (Fullerton, CA, USA). Anti-phospho-p38 and anti-p38 antibodies were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Whole cell lysates (50 µg) were boiled in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel loading buffer, subjected to SDS-PAGE, transferred to a nitrocellulose membrane, and probed with appropriate primary and peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Chemiluminescent signaling was developed using Super Signal West Pico or Femto substrate (Pierce, Rockford, IL, USA). The blots were imaged and band densities were quantified with a Gel Doc 2000 Chemi Doc system and Quantity One software (Bio-Rad, Hercules, CA, USA). Values were normalized to those of β-actin as the loading control.
Real-time polymerase chain reaction (PCR)
Total RNA from cells was isolated using the acid guanidinium thiocyanate-phenol-chloroform method. Complementary DNA was prepared from total RNA using the MAXIME RT Premix Kit (iNtRON Biotechnology Inc., Seongnam, South Korea). Real-time PCR was performed using the Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and the Super Script III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen, Carlsbad, CA, USA). The primers used for human VCAM-1 were as follows: (sense) 5'-CAT GGA ATT CGA ACC CAA ACA-3' and (antisense) 5'-GGC TGA CCA AGA CGG TTG TATC-3'. The primers used for human ICAM1 were as follows: (sense) 5'-GCC CTT TCT CGT CAT TTA GAT CTC-3' and (antisense) 5'-CT ATG TTA ATA TGA GCT TTG ACA AAA-3'. The primers used for human GAPDH were as follows: (sense) 5'-ATG GCA TCA AGA AGG TGG TG-3' and (antisense) 5'-CAT ACC AGG AAA ATG AGC TTG-3'. Dissociation curves were monitored to check for aberrant formation of primer dimers. The results were interpreted by the relative quantity method (ΔΔCt).
Monocyte-endothelial cell adhesion assay
HUVECs were seeded in 24-well plates until they reached a confluent monolayer and were incubated with NM (10 µg/mL) (Sigma, St. Louis, MO, USA) for 24 h in EGM-2 medium. Human recombinant tumor necrosis factor-α (TNF-α) was added to the appropriate wells (10 ng/mL) for 18 h before adding monocytes. Monocyte adhesion was quantified by counting the cells. Wells containing HUVECs without U937 cells were used as blanks.
Dihydroethidine (DHE) staining
Intracellular superoxide was detected using the superoxide-sensitive fluorophore DHE. Cells were grown in chamber slides (2×105 cells/well) (Nalgene Nunc International, Penfield, NY, USA). Cells were rinsed three times with 3 mL of Krebs-HEPES buffer and then incubated in 10 µM DHE for 20 min at 37℃ in the buffer. The DHE was washed from the cells to avoid the absorption of any extracellular oxyethidium formed by auto-oxidation, and the fluorescence intensity of DHE in the cells was imaged and measured using a confocal microscope.
Hydrogen peroxide measurements
Hydrogen peroxide produced by live cells was measured in HUVEC-conditioned medium using an Amplex Red Hydrogen Peroxide Assay Kit (Molecular Probes, Sunnyvale, CA, USA) as recommended by the manufacturer.
Statistical analysis
All experiments were performed at least three times. All data are expressed as means±standard deviations. Statistical analysis was performed using Sigma Stat (Systat Software, La Jolla, CA, USA). Data in which two conditions were compared were evaluated using one-way analysis of variance followed by Tukey's post-hoc test; p-values <0.05 were considered significant.
RESULTS
Effect of NM on endothelial cell viability
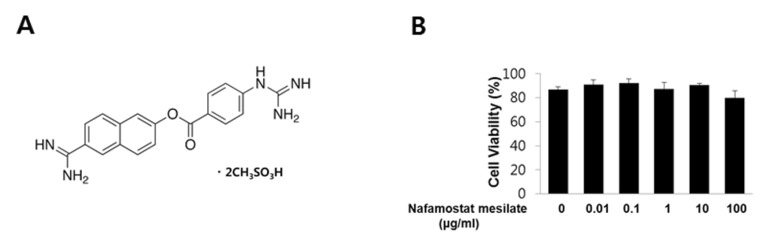
It is not known whether NM can induce vascular endothelial cell death. Therefore, we first investigated the effect of NM on endothelial cell viability. Fig. 1A shows the structure of NM. Endothelial cells were exposed to various concentrations of NM (0.01~100 µg/mL) for 24 h and then tested for viability using PI staining. As shown in Fig. 1B, NM did not affect endothelial cell viability.
NM inhibits TNF-α-induced VCAM-1 and ICAM-1 expression in HUVECs
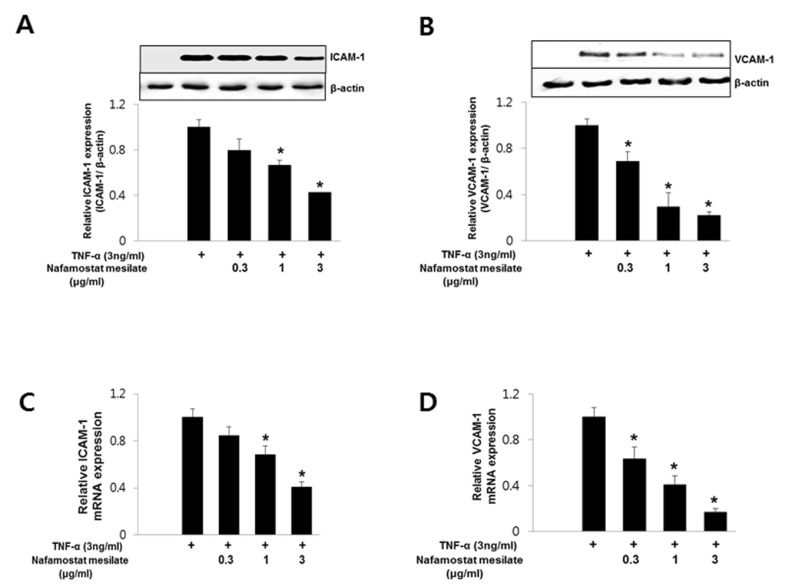
Next, we investigated whether NM affected VCAM-1 and ICAM-1 expression in endothelial cells in response to TNF-α. Endothelial cells were incubated with NM for 30 min before treatment with TNF-α (3 ng/mL). After treatment with TNF-α for 8 h, the endothelial cells were harvested for Western blot analysis and real-time PCR. VCAM-1 and ICAM-1 protein expression was not detected in un-stimulated endothelial cells, but treatment with TNF-α resulted in a marked increase in VCAM-1 and ICAM-1 protein expression. Pretreatment with NM at 0.3~3 µg/mL inhibited TNF-α-induced VCAM-1 and ICAM-1 protein expression dose-dependently in HUVECs (Fig. 2A and 2B). Similarly, VCAM-1 and ICAM-1 mRNA expression was induced by treatment with TNF-α, but the effect was decreased significantly by NM (Fig. 2C and 2D). These results suggest that NM acts as an inhibitor of TNF-α-induced VCAM-1 and ICAM-1 expression in endothelial cells.
NM reduces TNF-α-induced ROS production in HUVECs
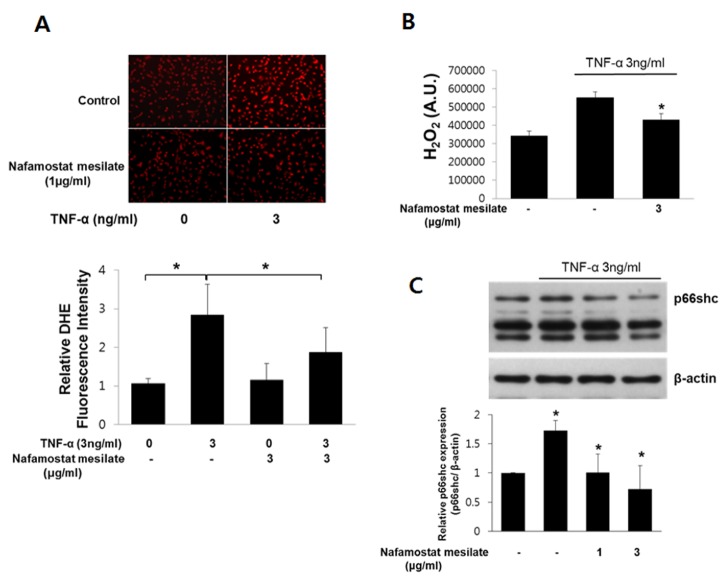
Vascular oxidative stress is a mechanism through which TNF-α induces vascular inflammation [15]. Therefore, we investigated whether NM would suppress intracellular ROS production induced by TNF-α in cultured human endothelial cells. ROS production was assessed using DHE staining and an Amplex Red Hydrogen Peroxide Assay Kit. TNF-α increased the ROS level significantly in endothelial cells; however, the increase in ROS production induced by TNF-α was suppressed by NM (Fig. 3A and 3B). These results suggest that NM acts as an antioxidant to limit TNF-α-induced ROS production. P66shc is a key protein in the regulation of intracellular ROS levels. It was previously shown that TNF-α increases ROS production by activating p66shc [16]. Thus, we next examined the relationship between TNF-α and p66shc activation, and the role of NM in TNF-α-induced p66shc activation in endothelial cells. We measured the p66shc expression level in endothelial cells following TNF-α treatment (Fig. 3C). Our results indicate that the TNF-α-stimulated increase in p66shc expression was significantly blunted in HUVECs treated with NM. These findings show that the antioxidant properties of NM in HUVECs may be due to the inhibition of p66shc activation.
NM suppresses TNF-α-stimulated p38 MAPK phosphorylation in HUVECs
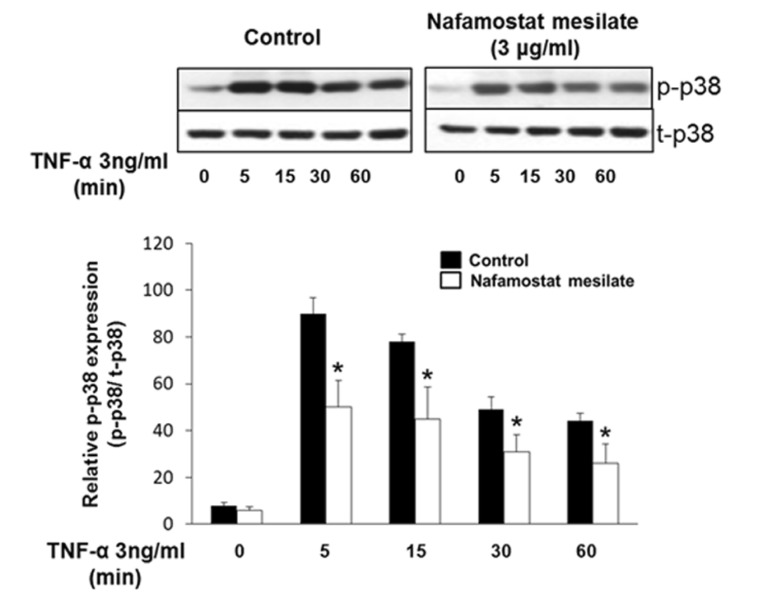
Next, we examined whether NM inhibits p38 MAPK phosphorylation stimulated by TNF-α in endothelial cells. As shown in Fig. 4, TNF-α induced p38 MAPK phosphorylation in HUVECs. However, this increase was markedly inhibited by NM.
NM suppresses monocyte adhesion in TNF-α-stimulated HUVECs
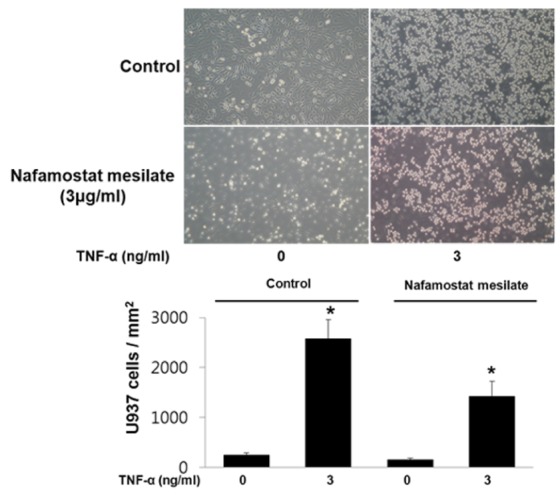
Adhesion molecules on endothelial cells induce monocyte adhesion to the vessel as well as vascular inflammation. Therefore, we were interested in whether NM regulates TNF-α-stimulated monocyte adhesion in endothelial cells. We examined the adhesion of U937 cells to TNF-α-stimulated HUVECs. As shown in Fig. 5, TNF-α stimulated the adhesion of U937 cells to HUVECs, whereas the TNF-α-stimulated adhesion of U937 cells was significantly diminished in HUVECs treated with NM.
DISCUSSION
In the present study, we investigated the effect of NM on vascular endothelial cell dysfunction induced by TNF-α and found that NM inhibited TNF-α-induced monocyte adhesion and the expression of endothelial cell adhesion molecules such as VCAM-1 and ICAM-1. We also found that the anti-adhesive effect of NM on endothelial cells was mediated through the inhibition of p66shc, ROS production, and p38 MAPK activation. This observation suggests that NM exerts protective effects against endothelial cell dysfunction.
NM inhibits the zymosan-stimulated production of superoxide, hydrogen peroxide, and hydroxyl radicals in human polymorphonuclear leukocytes [17]. Previously, we showed that the vasodilation of peripheral vessels and the endothelium protect against vascular endothelial cell apoptosis caused by NM [18]. This may be related closely to the function of NM in oxidative stress. In the present study, NM reduced TNF-α-induced ROS production in endothelial cells in vitro (Fig. 3A and 3B). These results suggest that NM acts as an antioxidant in TNF-α-induced ROS production.
Previous studies have demonstrated the important role that p66shc plays in ROS production in vascular endothelial cells and its involvement in endothelial dysfunction under pathophysiological conditions such as atherosclerosis [3,4]. In addition, p66shc acts as a key mediator of the TNF-α pathway, inducing endothelial cell dysfunction [16]; moreover, the production of ROS in osteoblasts in response to TNF-α exposure requires PCKβ/p66shc signaling [19]. The upregulation of p66shc was strongly associated with the expression of adhesion molecules, such as VCAM-1 and ICAM-1, and monocyte adhesion to endothelial cells. The stimulation of endothelial cells with TNF-α increased VCAM-1 and ICAM-1 expression, which was blocked markedly by the pretreatment of cells with NM (Fig. 1). Interestingly, NM abrogated the TNF-α-induced upregulation of p66shc and ROS production in endothelial cells (Fig. 3). Thus, our data suggest that p66shc is an important, but not the sole, mediator of endothelial cell dysfunction induced by TNF-α and that the antioxidant properties of NM in endothelial cells may be due to the inhibition of p66shc activation.
Not only p66sch but also p38 MAPK has an important role in vascular inflammation. The expression of p38 MAPK increases during the early stage of vascular inflammation and induces the upregulation of ICAM-1 expression in vascular endothelial cells [20]. This upregulation enhances the recruitment of leukocytes such as monocytes or basophils, which attack the vascular endothelium and create an atheroma and atherosclerotic changes in vessels. The upregulation of p38 MAPK in vascular endothelium mediates endothelial cell apoptosis through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway [21]. Our data demonstrate that TNF-α-induced p38 MAPK phosphorylation in endothelial cells was inhibited markedly by NM, suggesting that it has multiple anti-inflammatory effects on the vascular endothelium.
ROS and inflammatory cytokines induce p66sch and p38 MAPK upregulation in vascular endothelial cells and enhance VCAM-1 and ICAM-1 expression in vascular endothelial cell membranes. VCAM-1 and ICAM-1 play an important role in blood monocyte recruitment to the arterial intima during the early stages of atherosclerosis. In addition, previous studies have demonstrated the upregulation of VCAM-1 and ICAM-1 in experimental models of atherosclerosis [22]. In the present study, NM inhibited monocyte adhesion in TNF-α-stimulated endothelial cells (Fig. 5), suggesting that NM is a potent drug that can prevent monocyte recruitment and the development of atherosclerotic lesions.
In conclusion, our findings show that the inhibitory effects of NM on TNF-α-induced monocyte adhesion to endothelial cells are likely to have relevance for inhibiting not only VCAM-1 and ICAM-1 expression but also ROS production through decreased p66shc expression, which improves vascular endothelial cell function and prevents endothelial dysfunction related to vascular inflammation. NM may therefore represent a new pharmacological approach for the prevention and treatment of atherosclerosis.
 XML Download
XML Download