

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Long-term potentiation (LTP) is a long-lasting, activity-dependent enhancement of excitatory synaptic strength after application of a brief, high-frequency train of electrical stimulation [1,2]. Induction of LTP at pyramidal cell synapses in the CA1 region of the hippocampus requires the activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), metabotropic glutamate receptors (mGluRs), and N-methyl-D-aspartate receptors (NMDARs) [3]. Calcium/calmodulin-dependent protein kinase II (CaMKII), protein kinase C (PKC), and cAMP-dependent protein kinase A-three major serine/threonine protein kinases-have also been implicated in NMDAR-dependent LTP. In particular, PKC is involved in the induction of LTP in the hippocampus [4] and cerebellar Purkinje cells [5].
PKC plays an important role in transducing signals associated with a variety of cellular responses, including cell growth and differentiation, gene expression, hormone secretion, and membrane function. Ten PKC isoforms have been identified; these are divided into three classes based on sequence homology, substrate preference, and activators [6,7]. Activation of the classical PKC isoforms, α, β (splice variants βI and βII) [8] and γ, is significantly enhanced by calcium, diacylglycerol, and phorbol esters. Activation of the novel PKC isoforms, δ, ε, η and θ, is also enhanced by diacylglycerol and phorbol esters, but not by calcium. Activation of the atypical PKC isoforms, ξ and λ, is not influenced by calcium, diacylglycerol, or phorbol esters. Multiple PKC isoforms are expressed throughout the central nervous system, including the hippocampus [9-11], and can modulate ligand-gated ion channel function [12].
Recent studies examining the role of specific PKC isoforms in synaptic plasticity have identified protein kinase Mζ (PKMζ), an atypical isoform of PKC, as a candidate central player in maintaining late LTP. PKMζ maintains late LTP by persistently modifying N-ethylmaleimide- sensitive factor (NSF)/mGluR2-dependent AMPAR trafficking to favor receptor insertion into postsynaptic sites [13,14]. PKCγ, like the other classical PKC isoforms, PKCα and β, is activated by a receptor-coupled mechanism that mediates breakdown of inositol phospholipid, generating inositol 1,4,5-trisphosphate and the PKC-activating lipid, diacylglycerol. Several studies have shown that normal neuronal function, including LTP and LTD, requires PKCγ; for example, LTP is altered in the hippocampus of PKCγ-deficient mice [11].
Phorbol 12-myristate 13-acetate (PMA; also known as 12-O-tetradecanoylphorbol 13-acetate) is a stable analog of the signaling membrane lipid diacylglycerol and is thus a specific activator of PKC. However, PMA, the most commonly used phorbol ester, is a known tumor promoter that has been shown to act as a co-mitogen with other factors in a number of cell types, including fibroblasts [15]. PMA is also known to potentiate exocytosis and modulate vesicle fusion kinetics in neurons and endocrine cells [16]. PMA, acting through PKC, causes activation of extracellular signal-regulated kinase (ERK) and formation of new spines in cultured hippocampal neurons [17]. Therefore, the actions of PMA may reveal a role for PKC in NMDA receptor-dependent LTP in the hippocampus and, ultimately, in learning and memory. However, the specific mechanism underlying the actions of PMA is not clear, largely owing to the diversity of PKC isoforms. Thus, the purpose of this study was to identify which PKC isoforms are responsible for PMA-induced augmentation of LTP in the hippocampus.
METHODS
Experimental animals
Male, 3~5-wk-old C57BL/6N mice (Samtako, South Korea) were used in this study. All animals were individually housed in a temperature-controlled room (22~25℃) under a 12 h light/12 h dark cycle (lights on at 07:00 AM). Food and water were available ad libitum. All experiments were approved by the institutional Animal Care and Use Committee of Eulji University (permit no. EUIACUC 11-12).
Drugs
The role of PKC activation in the induction of LTP was examined using PMA, Ro 31-8220 (a nonspecific PKC inhibitor), rottlerin (a selective PKCδ inhibitor) and DL-APV (DL-2-amino-5-phosphonovaleric acid; NMRA receptor antagonist), obtained from Sigma Chemical Co. (St. Louis, MO, USA). The PKCε inhibitor, TAT-εV1-2 peptide, was from Anaspec (San Jose, CA, USA). All drugs were dissolved in dimethylsulfoxide (DMSO) except for DL-APV, which was dissolved in normal saline. All drugs were stored at -20℃ prior to use in experiments.
Preparation of hippocampal slices for extracellular recording
Mice were decapitated under deep enflurane anesthesia and brains were quickly removed and transferred to ice-cold dissection buffer containing sucrose (212.7 mM), KCl (2.6 mM), NaH2PO4 (1.23 mM), NaHCO3 (26 mM), dextrose (10 mM), MgCl2 (10 mM), and CaCl2 (0.5 mM). Horizontal brain sections (400µm in thickness), prepared using a vibratome (Campden Instruments, Loughborough, UK), were placed into dissection buffer that was continuously bubbled with 5% CO2/95% O2 (v/v). The slices were held at 35℃ for 1 h in a chamber filled with continuously oxygenated artificial cerebrospinal fluid (ACSF) with the following composition: NaCl (124 mM), KCl (5 mM), NaH2PO4 (1.25 mM), NaHCO3 (26 mM), dextrose (10 mM), MgCl2 (1.5 mM), and CaCl2 (2.5 mM). The slices were then transferred to an open submersion-type recording chamber, maintained at 30℃, and perfused with oxygenated ACSF at a flow rate of 2 ml/min.
Electrophysiological recording
A bipolar stimulating electrode was inserted into the stratum radiatum to activate the Schaffer collaterals of CA1 hippocampal pyramidal neurons. A glass micropipette filled with ACSF was inserted into the CA1 pyramidal layer to record field excitatory post-synaptic potentials (fEPSPs). fEPSPs in the CA1 layer were evoked by stimulating Schaffer collaterals with a 0.2 ms electrical pulse delivered through concentric bipolar stimulating electrodes (FHC; Bowdoinham, ME, USA). The initial slope of extracellular fEPSPs was recorded in the CA1 stratum radiatum. Baseline responses were obtained upon application of a 50%-maximal-intensity stimulus at 0.033 Hz. LTPs were induced using a conventional theta burst stimulation (TBS) protocol consisting of eight bursts, each composed of four 100-Hz pulses, administered at 200-ms intervals. The stimulus intensity during TBS was identical to that of the test pulse. All measurements are expressed as percentages of average values calculated 20 min prior to LTP induction. Differences between groups were determined by comparing average LTP values 58~60 min after LTP induction. To measure paired-pulse facilitation (PPF), we used interstimulation intervals (ISIs) of 25, 50, 100, 200, 400, 1,000, and 2,000 ms.
Statistics
Data were analyzed using SPSS version 10.0 software (SPSS Inc., Chicago, IL, USA). All values are given as means±SEMs. Statistical significance was assessed using Student's t-test or analysis of variance (ANOVA) followed by Fisher's PLSD (protected least-significant difference) post hoc test, as appropriate.
RESULTS
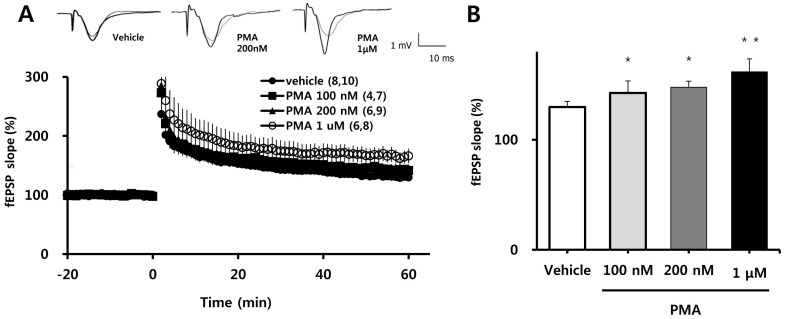
PMA enhances the induction of LTP
Because four episodes of TBS induce very robust LTP, effects of PMA on the magnitude of LTP might not be detectable under such conditions. Therefore, we measured the magnitude of LTP induced by TBS episode 1, 2, 3 and 4 and selected the episode that induced the lowest magnitude LTP. Next, we tested the effects of different concentrations of PMA on LTP 60 min after induction of LTP by one episode of TBS. PMA was applied to the hippocampal slice at least 1 h prior to application of TBS. PMA did not affect the magnitude of baseline fEPSP values, but as shown in Fig. 1A, it exerted a concentration-dependent potentiation of TBS-induced LTP. This is quantified in bar graph form in Fig. 1B, which shows stepwise percentage increases in average LTP values 58~60 min after one episode of TBS (relative to baseline) with application of increasing concentrations of PMA (100 nM: 142±4.38% ; 200 nM: 146±5.51%, p<0.05; 1µM: 164±11.14%, p<0.01) compared to vehicle (130±5.15%).
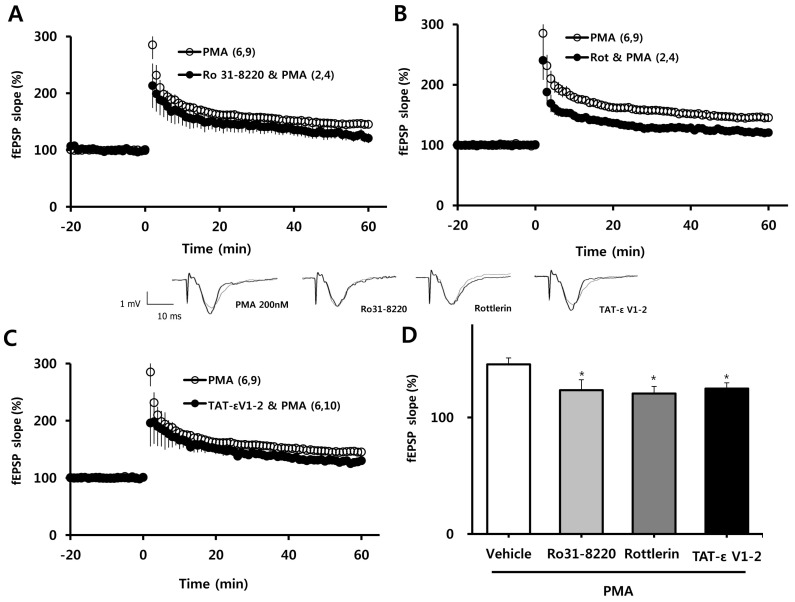
Inhibition of PKCδ or PKCε blocks the facilitation of LTP by PMA
We next determined which PKC isoform was involved in the facilitating effect of PMA (200 nM) on the induction of LTP. PMA facilitation of LTP was blocked in the presence of the nonspecific PKC inhibitor, Ro 31-8220 (10µM; Fig. 2A); the selective PKCδ inhibitor, rottlerin (1µM; Fig. 2B); and the selective PKCε inhibitor, TAT-εV1-2 peptide (500 nM; Fig. 2C). These results are quantified in bar graph form in Fig. 1D, which shows the average LTP values 58~60 min after TBS (as a percent of baseline) in slices pretreated with PMA plus 10µM Ro 31-8220 (123±9.1%), PMA plus 1µM rottlerin (121±6.0%), or PMA plus 500 nM TAT-ε V1-2 peptide (125±4.9%) compared to PMA alone (146±5.5%). Collectively, these data indicate that PMA exerts its potentiating effect on TBS-induced LTP in the hippocampal CA1 region via activation of PKCδ or PKCε.
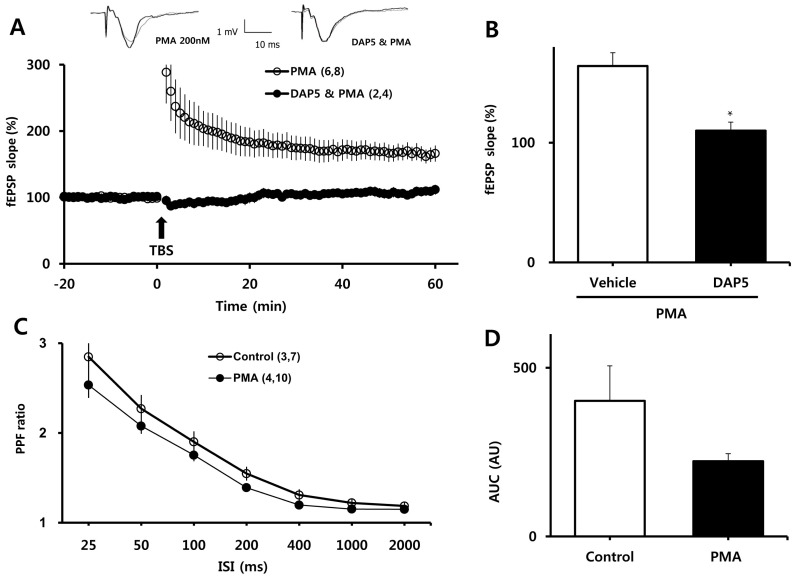
PMA-mediated facilitation of LTP induction requires NMDAR activity
To examine the involvement of NMDARs in PMA-induced enhancement of LTP, we measured LTP in the presence of DL-APV, an NMDAR blocker. As shown in Fig. 2A, B, DL-APV (50µM) eliminated the enhancing effect of PMA on LTP (110±7.13% vs. 164±11.14% of baseline for DL-APV and PMA, respectively; p<0.001). We then examined whether the facilitating effect of PMA was attributable to changes in basal synaptic transmission. PPF ratios, reflecting presynaptic function, were determined before and at least 30 min after treatment with PMA (Fig. 3C). PPF ratios were similar between the PMA-treated group and the vehicle-treated group, although there was tendency toward lower PPF ratios in slices treated with PMA.
DISCUSSION
In the present study, we showed that PMA dose-dependently facilitates the induction of hippocampal LTP and exerts this effect through PKCδ or PKCε. Moreover, this potentiating effect requires activation of NMDARs.
Because of its high sensitivity to intracellular concentrations of calcium, PKC is thought to be a major coordinator of processes underlying activity-induced synaptic modifications. Consistent with this role, PKC activation is necessary for maintenance of LTP in cultured neurons [18] and hippocampal slices [19,20]. In the present study, we have shown that activation of PKC also enhances the induction of LTP, an effect that may be attributable to NMDAR trafficking. Activation of PKC increases the NMDA channel opening rate and, importantly, delivers new NMDARs to the plasma membrane [21,22]. Recently, it has been shown that PKC promotes NMDA receptor trafficking and induces synaptic plasticity by indirectly triggering autophosphorylation of CaMKII, which subsequently becomes increasingly associated with NMDARs [23]. PKC also regulates NMDAR function by phosphorylating the glutamate receptor subunit NR1 and promoting export and slow transport of NMDARs from the endoplasmic reticulum to the plasma membrane over a period of hours [24,25] and by interacting with NMDAR-associated proteins and promoting the rapid insertion NMDAR channels in hippocampal neurons [22,26]. Alternatively, PKC activation may enhance LTP due to synaptogenesis or phosphorylation or insertion of AMPARs into the postsynaptic membrane, thereby altering the sensitivity of the membrane to glutamate [27,28]. In addition, the PKC activator, bryostatin-1, has been shown to increase the number of dendritic spines [29,30].
In CA1 pyramidal cells, PKC activation has also been shown to induce memory-specific enhancement of synaptic potentials elicited by Schaffer collateral stimulation [31]. Application of exogenous compounds known to activate PKC, such as oleic acid [32], arachidonic acid [33] or glucose [34], synergistically enhance the induction of LTP, converting short-term potentiation to LTP [32,33]. Some PKC activators, such as bryostatin-1, enhance memory-specific increases in the number of mushroom spines, which provide structural storage sites for long-term associative memory and sites for memory-specific synaptogenesis that involve PKC-regulated changes in spine shape as well as PKC-regulated changes in pre- and postsynaptic ultrastructure [35]. Our demonstration that activation of PKC by PMA regulates the induction of LTP could support suggestions by previous studies that PMA regulates a structural change in spines during memory retention [36-39].
We have shown that enhancement of LTP by PMA requires activation of PKCδ or PKCε. PMA has been previously shown to modulate both the extent and fusion kinetics of vesicle exocytosis and to facilitate exocytosis and vesicle fusion by activating PKCα in neuroendocrine PC12 cells [16]. Moreover, PMA potentiation of NMDA-induced currents in primary cultured cerebellar granule cells is mediated by protein kinase Cα [40]. Also, PMA stimulates neutrophil adhesion to endothelial cells in association with PKCδ translocation to the plasma membrane [41]. Contrasts between literature reports and the results of the present study may be attributable to differences in cell types or species studied [42]. However, we did not evaluate the involvement of other PKC isoforms, perhaps also activated by PMA, in the induction of LTP. Thus, we cannot exclude the possibility that such isoforms also facilitate LTP induction by PMA.
In conclusion, our results suggest that PMA regulation of synaptic efficacy and synaptogenesis may be mediated by activation of PKCδ or PKCε via a mechanism that depends on NMDAR activity.
 XML Download
XML Download