

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Atopic dermatitis (AD) is an inflammatory, chronically relapsing, non-contagious and pruritic skin disorder characterized by allergic symptoms such as redness, inflammation, and itching [1]. More studies of anti-atopic agents must be conducted because the exact pathogenesis of AD is unknown and a definitive treatment is lacking.
Taxifolin 3-O-β-D-glucopyranoside (taxifolin glycoside) is a flavonoid compound isolated from Rhododendron mucronulatum, which has been used for treatment of rheumarthritis, neuralgia, and inflammation traditional oriental medicine [2,3]. Recent studies indicate that taxifolin glycoside inhibits dendritic cell-mediated immune response and demonstrates anti-inflammatory activity in NC/Nga mice: an atopic dermatitis model [4,5]. In a therapeutic efficacy study in NC/Nga mice with AD-like lesions, a rapid and profound restoration of skin barrier function was observed in the group treated with the taxifolin glycoside-loaded Pep-1 peptide-conjugated elastic liposomes (Pep1-EL) formulation [6]. So far, taxifolin glycoside has been tested as a potential therapeutics for AD.
The K+ channel controlled by the human ether-a-go-go related gene (hERG) conducts the rapidly activating delayed rectifier K+ current (Ikr) in cardiac myocytes, which plays a central role in the repolarization of action potentials [1-3]. Loss of function or blockade of the hERG K+ channel can induce long QT syndrome (LQTS), which is characterized by a prolonged QT interval on an electrocardiogram and can result in fatal arrhythmias such as torsade de pointes [7,8]. Many drugs have reported side effects, including hERG K+ current inhibition, which can lead to acquired LQTS [9-12]. However, little information is available regarding the anti-arrhythmic effect and hERG K+ channel blockade that are caused by taxifolin glycoside. In this study, we tested the cardiovascular safety of taxifolin glycoside by exmining its effects on hERG K+ channels using a whole cell patch clamp method.
METHODS
Cell cultures
hERG cDNA was a generous gift from Dr. Tong Mook Kang, Ph.D. (Professor, Department of Physiology, College of Medicine, Sungkyunkwan University, Suwon, Korea). hERG cDNA was subcloned into mammalian expression vector pCDNA3.1 (Invitrogen) for transfection into cells. Transfection of hERG into Chinese Hamster Ovary (CHO) cells was achieved via the lipofectamin method (Life Technologies). The stably transfected hERG-CHO cells were generated through G418 (Gibco) antibiotic selection. The transfected clones were selected through the use of 1,000 µg/ml of G418. The CHO cells stably expressed hERG (hERG-CHO) were maintained in F-12 supplemented with 10% (v/v) fetal bovine serum (FBS) and 500 µg/ml G418. For electrophysological recordings, the cells were harvested from the culture dish via trypsinization and were seeded onto a glass coverslip.
Taxifolin glycoside
Taxifolin glycoside was kindly provided from Dr. Min Won Lee, Ph.D. (Professor, College of Pharmacy, Chung-Ang University, Seoul, Korea) and was dissolved in distilled water to obtain a 500 mM stock solution, which was stored at -20℃. Taxifolin glycoside was diluted in external solution to achieve the final concentration.
Electrophysiological recordings
The hERG K+ currents were recorded using the whole-cell patch clamp technique. Cells on a coverslip were transferred to a recording chamber mounted on the stage of an inverted microscope (Olympus). For the hERG K+ current recording, the bath was perfused with HEPES-buffered Tyrode solution containing (in mM) NaCl 155, KCl 5, MgCl2 1, HEPES 10, and glucose 10 (pH 7.3 with NaOH). The bath solution was perfused at a rate of 0.7 ml/min. The pipette solution contained (in mM) KCl 140, Mg-ATP 5, MgCl2 1, KH2PO4 10, EGTA 0.1, and HEPES 5 (pH 7.2 with KOH). The electrodes were constructed from borosilicate glass (WPI) using a micropipette puller (Narishige) and were heat-polished with a microforge (Narishige). The resistance of the electrode when filled with the pipette solution was 2~3 MΩ. Membrane currents were recorded with an Axopatch 200B amplifier (Axon Instruments) and a Digidata 1322A (Axon Instruments). Currents were filtered at 2 kHz and digitized at 10 kHz. Capacitance and series resistance compensations were optimized. The pCLAMP 9.2 software (Axon Instruments) was used to generate voltage clamp protocols, acquire data, and analyze current traces.
Statistical analysis
Data were expressed as mean±standard error of the mean (SEM). Data analysis and curve fitting were performed with Clampfit 9.2 (Axon Instruments) and Origin 8.0 software (OriginLab). Statistical comparisons were made using a paired t-test. p-values<0.05 were considered statistically significant.
RESULTS
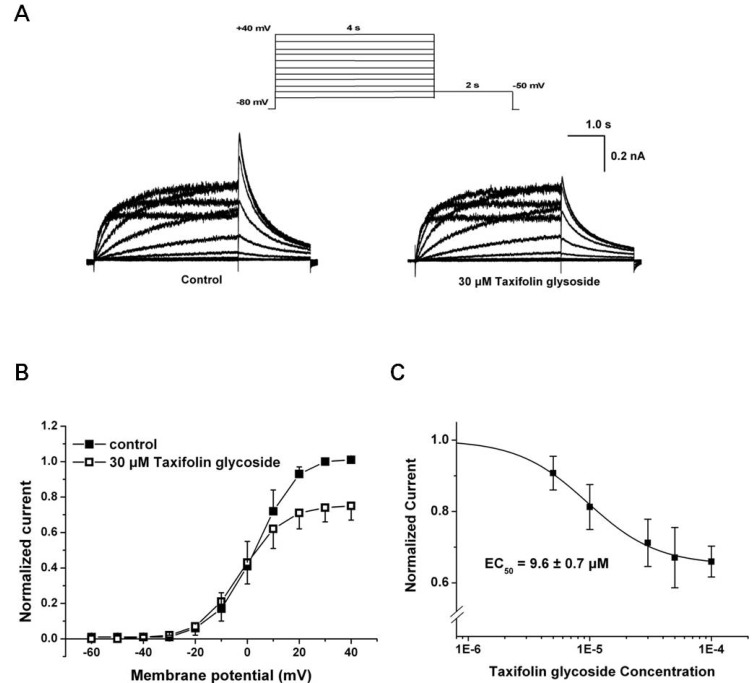
We investigated the effect of taxifolin glycoside on hERG K+ current stably expressed in CHO cells using the whole-cell patch clamp technique. The hERG K+ currents were elicited from a holding potential of -80 mV by the test pulses ranging from -60 mV to +40 mV in 10 mV steps (Istep). Each test pulse had 4 seconds duration and was followed by a repolarization step to -50 mV for 2 seconds to evoke tail current (Itail). The hERG K+ currents were first recorded under control conditions, and then 30 µM taxifolin glycoside was added into the bath for 20 minutes. Taxifolin glycoside reduced tail currents of hERG K+ channels during the repolarizing pulse (Fig. 1A). Normalized current-voltage (I-V) relationships of Itail are illustrated in Fig. 1B (n=5), which shows that Itail was increased with depolarizing voltages and reached a plateau at approximately +30 mV. Taxifolin glycoside reduced Itail amplitudes after a test pulse to 0 mV.
At taxifolin glycoside concentrations of 5, 10, 30, 50 and 100 µM, the peak amplitude of the Itail were measured in the same cell and were normalized to the control value. Taxifolin glycoside blocked Itail in a concentration-dependent manner. To obtain the half maximal effective concentration (EC50) of taxifolin glycoside, the concetrtion-response relationships of Itail were fitted using the Hill equation. The EC50 of taxifolin glycoside was 9.6±0.7 µM (n=5, Fig. 1C). The fractional blockages in Itail were 12.0±2.5%, 18.1±4.7%, 26.3±4.5%, 35.4±5.6% and 36.9±3.0% in the 5, 10, 30, 50 and 100 µM taxifolin glycoside groups, respectively (n=5). The effect of taxifolin glycoside was irreversible (data not shown).
To further understand the inhibitory effect of taxifolin glycoside on hERG K+ channels, we investigated the changes in channel kinetics induced by taxifoline glycoside.
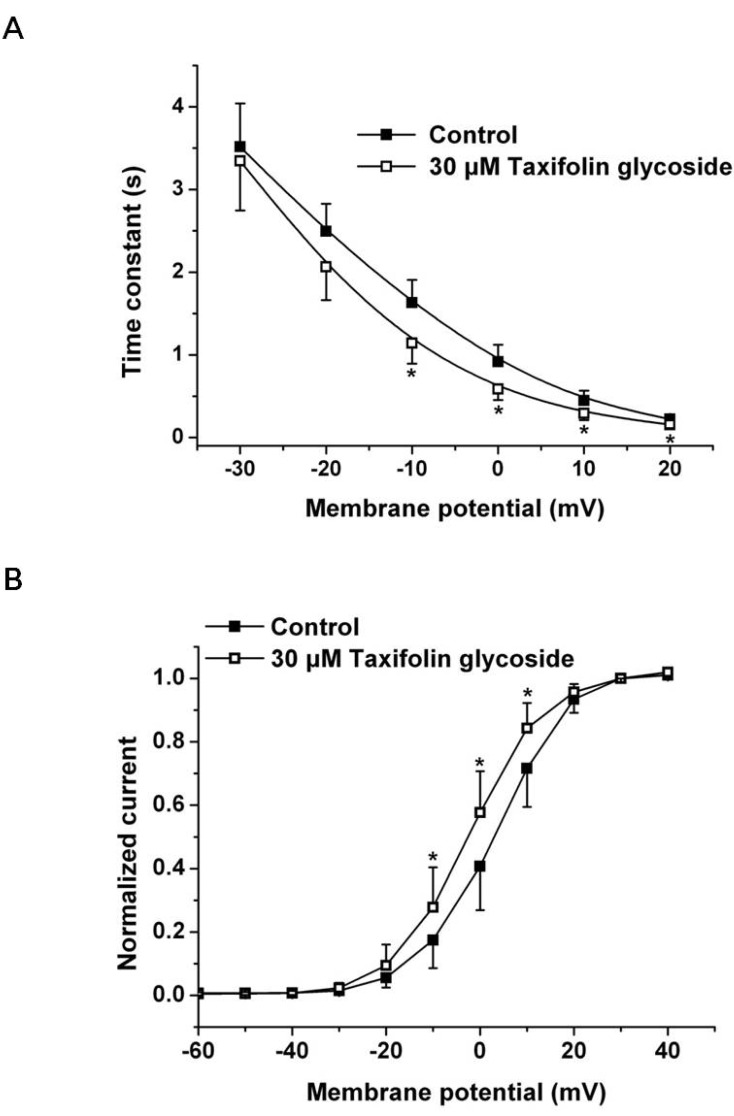
We first examined the effects of 30 µM taxifolin glycoside on the time constants of activation and the activation curve. To obtain the time constant of activation, the increasing phase of Istep in Fig. 1A was fitted to a single exponential function for test potentials in the range of -30 mV to +20 mV. The time constant of activation was significantly smaller in the presence of 30 µM taxifolin glycoside and their time constants were reduced by 29.9±4.3% at +10 mV (n=5, p<0.05, Fig. 2A). Relative to the control, hERG K+ currents after taxifolin glycoside treatment activated more rapidly. And the activation curve was constructed by normalizing the Itail shown in Fig. 1A. The normalized data were plotted against the voltage and fitted with the Boltzmann function (Fig. 2B). Taxifolin glycoside significantly changed the half-maximal activation voltages (V1/2: 2.7±2.1 mV and -2.2±0.1 mV; in the control group and in the group treated with 30 µM taxifolin glycoside, respectively; n=5, p<0.05). A 30 µM concentration of taxifolin glycoside produced a significant negative shift of the activation curve.
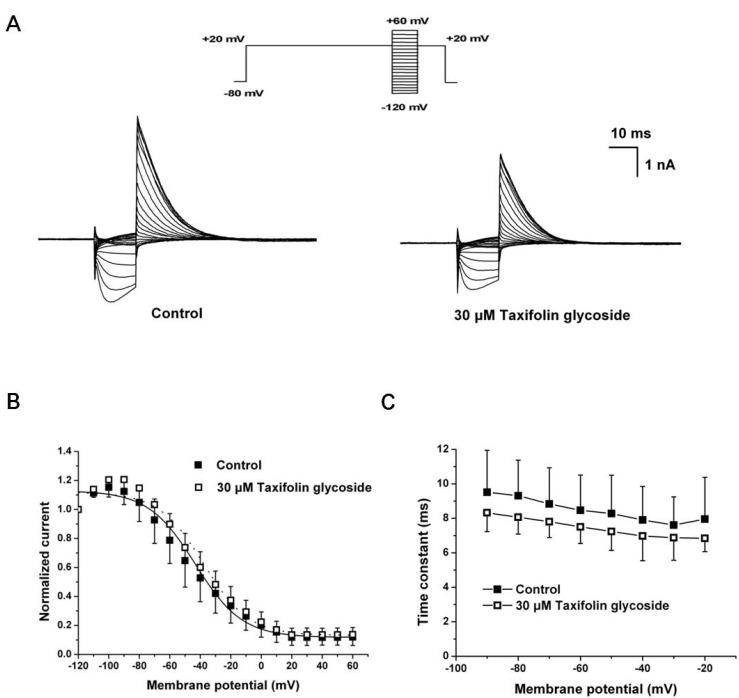
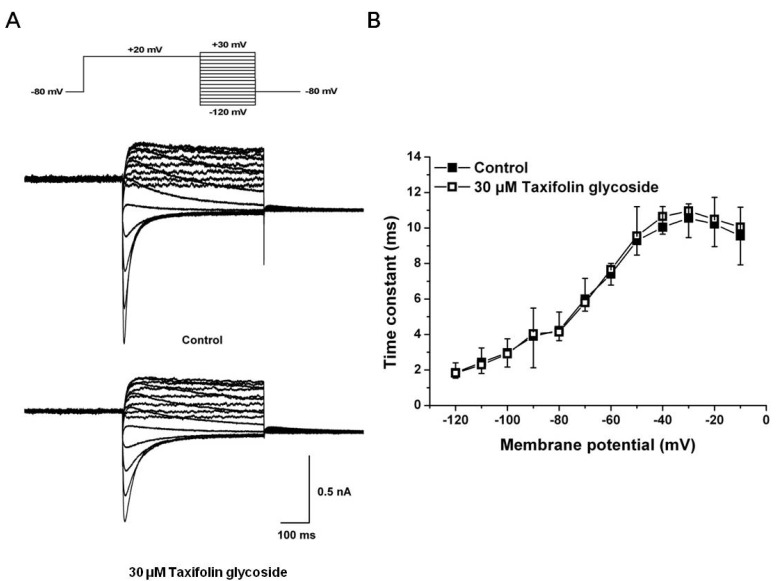
We next examined the effects of taxifolin glycoside on the time constants of steady-state inactivation, onset of inactivation and recovery from inactivation. To measure steady-state inactivation, a special protocol was used to inactivate the channel at a holding potential of +20 mV for 2 seconds, recover the channel from inactivation at various potentials ranging from -120 mV to +60 mV for 15 seconds in 10 mV steps, and then measure the resulting peak outward current at a constant +20 mV in the absence and the presence of 30 µM taxifolin glycoside (Fig. 3A). In Fig. 3B, the inactivation outward current amplitude was normalized and plotted against the test pulse potential, producing the steady-state inactivation curve. This curve was fitted with a Boltzmann function. Taxifolin glycoside did not affect the half-maximal inactivation values of hERG K+ channels (V1/2: -43.3±5.4 mV and -37.8±7.4 mV; in control and after treatment with 30 µM taxifolin glycoside, respectively; n=5). The time constant of channel inactivation was also assessed. To obtain the time constants of steady-state inactivation, the decays of the outward current in Fig. 3A were fitted to single exponential functions and plotted against voltages (Fig. 3C). Although taxifolin glycoside caused a slight decrease in the time constants of direct channel inactivation, the changes were not statistically significant (n=5, p>0.05).
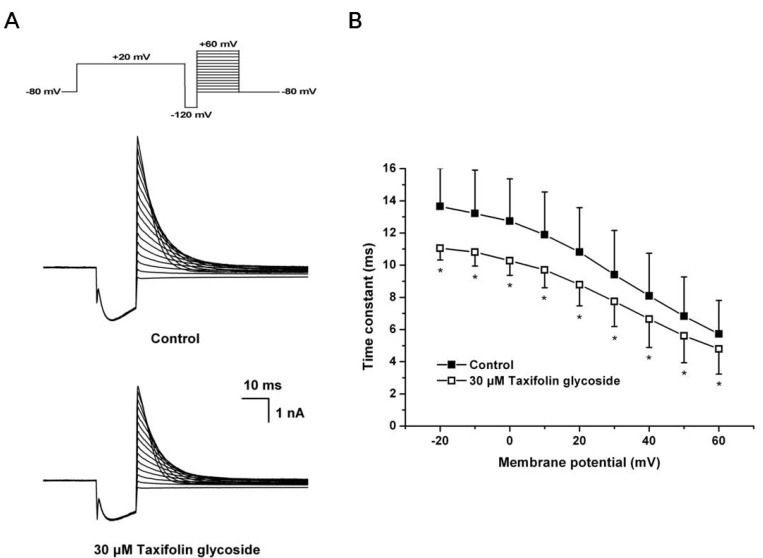
A 3-pulse protocol was used to assess the onset rate of channel inactivation. The channels were first inactivated by clamping the membrane at +20 mV for 2 seconds, followed by a prepulse to -120 mV for 15 microseconds. This prepulse was sufficiently long to allow for the rapid recovery of channels from inactivation, but short enough to prevent significant channel deactivation. Following the recovery prepulse, a series of test pulses were delivered with potentials ranging from -80 mV to +60 mV, resulting in large, outward inactivating currents. The current traces at the onset of inactivation in the absence and the presence of 30 µM taxifolin glycoside are shown in Fig. 4A. The time constants for the onset of inactivation were obtained by fitting a single exponential function to the decaying current traces during the third pulse of the protocol. The time constants of onset of inactivation were significantly decreased in the presence of 30 µM taxifolin glycoside (n=5, p<0.05, Fig. 4B).
The fully activated I-V protocol was used to determine the recovery from inactivation. Each current was obtained through depolarization to +20 mV for 2 seconds to inactivate the hERG K+ channels, followed by varying the repolarizing pulses to test potentials between -120 mV and +30 mV in 10 mV steps. The prepulse potential at +20 mV was sufficiently positive to induce full conductance of the hERG K+ channels but also rendered a large number of the channels inactive. The current traces for recovery from inactivation in the absence and the presence of 30 µM taxifolin glycoside are shown in Fig. 5A. The increasing phase of the tail current represents the rapid recovery from inactivated states to open states. The time constant for recovery from inactivation were obtained by fitting a single exponential function to the initial increase in tail current amplitude (n=5, Fig. 5B). Taxifolin glycoside did not affect the time constants for recovery from inactivation.
DISCUSSION
The present study demonstrated that taxifolin glycoside directly blocks hERG K+ channels in hERG-CHO cells. Taxifolin glycoside induced 2 distinct changes in hERG K+ channel gating: (1) facilitating activation (negative shift of the voltage-dependence of activation and accelerated activation time constant) and (2) facilitating of inactivation (reduced onset of inactivation time constant).
In Fig. 1, the blockage by taxifolin glycoside was significantly increased at +30 mV at which point the hERG K+ channel was fully activated. These results suggest that blockage by taxifolin glycoside might occur via open-state blockage. In addition, the hERG K+ channel kinetics related to activation were changed. Taxifolin glycoside produced a significant negative shift of the activation curve, and the time constant of activation was significantly decreased in the presence of taxifolin glycoside (Fig. 2). These data indicate that the activation of hERG K+ channels was accelerated by taxifolin glycoside and that taxifolin glycoside may block the hERG K+ channel when it is open. We also investigated the effects of taxifolin glycoside on hERG K+ channel inactivation kinetics [13]. Taxifolin glycoside significantly decreased the time constants for onset of inactivation (Fig. 4), although the properties of steady-state inactivation and recovery from inactivation were not changed (Fig. 3 and Fig. 5). These results indicate that the onset of inactivation of hERG K+ channels was accelerated by taxifolin glycoside and that taxifolin glycoside may also exhibit affinity for the inactivated state.
Because hERG K+ channel blockade has the potential to cause extensive side effects, in addition to LQTS and arrhythmias, in the process of developing new pharmaceutical drugs, the test for drug-induced hERG K+ channel blockade should be conducted prior to initiating in vivo toxicology studies [14]. Therefore, an understanding of drug-hERG K+ channel interaction at the molecular level is necessary for rational drug design. With regard to the relationship among hERG K+ current blockade, QT prolongation and fatal arrhythmia, Redfern et al. recommended a 30-fold safety margin between the maximum effective therapeutic plasma concentration of a drug and the IC50 of hERG [15]. In this study, we found that taxifolin glycoside directly inhibits hERG K+ currents in a concentration-dependent manner, with an EC50 of 9.6±0.7 µM. However, taxifolin glycoside of high concentration (more than 100 µM) did not produce further decrease of hERG K+ currents. Fig. 1C shows an unusual situation where the inhibition curve plateaus well above the control values. In this case, EC50 refers to relative IC50 which define the half concentration between the top and bottom plateaus of the curve [16]. Based on this data, the maximum therapeutic plasma concentration of taxifolin glycoside should be ≤0.32 µM. Recently, taxifolin glycoside has demonstrated anti-inflammatory and immune modulatory effects at concentrations between 10 and 20 µM in in vitro experiments [4-6]. However, the concentrations that are effective in vitro are higher than the recommended therapeutic plasma concentrations. Taxifolin glycoside also has the possibility to induce a cardiac arrhythmia, although the maximum inhibitory effects on hERG K+ currents was 36.9±3.0%, and taxifolin glycoside concentrations ≥100 µM did not produce a further dcrease of hERG K+ currents. Since outward K+ currents play an important role during plateau repolarization and in determining the configuration of the action potential, small changes in conductance can significantly alter the effective refractory period, hence the action potential duration [17]. So, if we use taxifolin glycoside as a topical ointment to treat inflammatory skin lesions such as atopic dermatitis and contact dermatitis, such treatment would be unlikely to produce side effects because the transcutaneous absorption rate is ≤1% that of oral delivery or injection.
In conclusion, taxifolin glycoside inhibited hERG K+ currents that function by facilitating activation and inactivation process. Our results provide an understanding of the cardiovascular risk profile of the new drug taxifolin glycoside.
 XML Download
XML Download