

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
ANOVA
analysis of variance
ASA
aspirin-treated group
DAB
diaminobenzidine tetrahydrochloride
DMSO
dimethyl sulfoxide
DPX
distrene plasticizer xylene
EtOH
ethanol
GFAP
glial fibrillary acidic protein
NSAID
non-steroidal anti-inflammatory drug
PB
phosphate buffer
SE
status epilepticus
SEM
standard error of the mean
TLE
temporal lobe epilepsy
Veh
vehicle-treated group
INTRODUCTION
Epilepsy is a chronic neurological disorder that can induce abnormal behavioral changes as well as neuronal dysfunction. Among various kinds of epilepsies, temporal lobe epilepsy (TLE) has the highest prevalence in humans [1]. Therefore, many studies have been conducted in order to identify its pathogenesis and develop therapeutic interventions. For TLE animal models, chemoconvulsants, including kainic acid (a glutamate analog) and pilocarpine (a muscarinic receptor agonist), have been employed [2]. It is well-known that kainic acid- or pilocarpine-induced status epileptics (SE) leads to neuronal cell death, gliosis, increased abnormal neurogenesis, and mossy fiber sprouting in the hippocampus [3-5]. Additionally, these events are consistently observed in patients with TLE. Epileptic insults induced by kainic acid or pilocarpine produce various cellular and molecular alterations, such as activation of ionotropic and metabotropic glutamate receptors as well as neurotrophin receptors, and changes in intracellular signaling systems [6-8]. However, the underlying events responsible for epileptogenesis remain unclear. Recently, it has been suggested that activation of pro-inflammatory signals may play a role in epileptogenesis [9]. For example, interleukin-1β signaling is chronically activated in tissue from both chronic epileptic rats and TLE patients [10]. In addition, interleukin-6 (IL-6) serum levels in epileptic patients are significantly higher compared to healthy controls and IL-6 was reported to be responsible for seizure development following viral infection [11,12]. Furthermore, inflammation induced by lipopolysaccharide deteriorated epileptic outcomes in SE animal models induced by lithium-pilocarpine or rapid kindling [13]. In accordance with these findings, it has been reported that anti-inflammatory agents could have favorable effects on epilepsy [14-16].
Aspirin (acetylsalicylic acid) is a typical non-steroidal anti-inflammatory drug (NSAID) [17]. Pharmacological actions of aspirin include anti-inflammatory, analgesic, anti-pyretic, and anti-thrombotic actions. Aspirin reduces acute myocardial infarction in the unstable angina [18]. In addition, daily aspirin treatment has been reported to diminish long-term risk of death from cancer [19] and to prevent cardiovascular diseases such as ischemic stroke [20]. Interestingly, the effect of aspirin on seizure/epilepsy is quite controversial. For example, in the kainic acid-induced or pilocarpine-induced SE model, salicylate, an active metabolite of aspirin, increased susceptibility and neuronal cell death [21,22]. However, aspirin treatment decreased neuronal loss in the hippocampal subfields in the lithium-pilocarpine-induced SE model [23]. In addition, aspirin dose-dependently reduced seizure incidence in the pentylenetetrazole-induced model, but not in the maximal electroshock model [24]. Therefore, considering the widespread use of aspirin to control fever, pain and inflammation, as well as its various prophylactic purposes, it is urgent to elucidate the effect of aspirin on seizure and epileptogenesis. The present study was designed to investigate the effect of aspirin on seizure susceptibility and hippocampal neuropathological changes following pilocarpine-induced SE.
METHODS
Chemicals
Aspirin, diaminobenzidine tetrahydrochloride (DAB), dimethyl sulfoxide (DMSO), distrene plasticizer xylene (DPX), pilocarpine hydrochloride, potassium permanganate, and terbutaline hemisulfate salt were purchased from Sigma-Aldrich (St. Louis, MO, USA) and atropine methyl nitrate was from Tokyo Chemical Industry Co. (Tokyo, Japan). Fluoro-Jade was purchased from Histo-Chem Inc. (Jefferson, AR, USA). Normal goat serum, goat anti-mouse IgG, and anti-rabbit IgG were purchased from Vector Laboratories (Burlingame, CA, USA). The glial fibrillary acidic protein (GFAP) antibody and Iba-1 antibody were from Chemicon International Inc. (Temecula, CA, USA) and Wako Pure Chemicals Inc. (Osaka, Japan), respectively. All other chemicals were purchased as the highest grade.
Experimental animals
Male C57BL/6 mice (aged 8 weeks, Koatech, Kyungki-do, Korea) were housed at standard temperature (22±1℃), humidity (50±1%), in a light-controlled environment (lights on from 8:00 a.m. to 8:00 p.m.) with ad libitum access to food and water. The Ethics Committee of The Catholic University of Korea approved all animal experiments and they were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23, revised 1996). Efforts were made to minimize animal suffering and reduce the number of animals used.
Drug administration
Aspirin was dissolved in 100% DMSO, and diluted in saline immediately before injection. According to the following formula [25], aspirin dosage was determined to satisfy that 15 or 150 mg/kg aspirin in a 23 g mouse is equivalent to a low dose of 100 mg or high dose of 1,000 mg in a 70 kg human, respectively.
[Interspecies scaling relationship: Dose of aspirin in mghuman = Dose of aspirin in mganimal (Weight of human in kg/Weight of animal in kg)0.7]
Aspirin- or vehicle- (10% DMSO in saline) treated mice were intraperitoneally (i.p.) injected daily for 10 days, starting from 3 days before the injection of pilocarpine and continuing until 6 days after the onset of SE.
Pilocarpine-induced status epilepticus model
Mice were administered atropine methyl nitrate (2 mg/kg, i.p.) and terbutaline hemisulfate salt (2 mg/kg, i.p.) 30 min before the injection of pilocarpine hydrochloride (280 mg/kg, i.p.) to minimize peripheral side effects. After pilocarpine administration, mice behavior was closely monitored for approximately 6 h to evaluate the onset time of first seizure, SE, severity, and mortality. The seizure stage was determined according to the Racine scale [26]: stage 1, facial clonus; stage 2, head nodding; stage 3, forelimb clonus; stage 4, rearing; and stage 5, rearing and falling. Animals that had stage 5 generalized tonic-clonic seizures (rearing and falling) were considered to show SE and were selected for further study. After 2 h of SE, diazepam (10 mg/kg, i.p.) was administered to terminate seizure. To facilitate recovery, all experimental animals were intraperitoneally injected with a 5% glucose solution, provided water-moistened chow, and housed in an incubator (30±1℃) for 5 days to maintain their physiologic body temperature. Mice were sacrificed 7 days after SE.
Sample preparation
Mice were anesthetized using 15% chloral hydrate and transcardially perfused with saline, followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4). After brains were quickly removed, they were cryoprotected with 30% sucrose solution for 3 days. Next, samples were quickly frozen with liquid nitrogen. Serial sections (20-µm-thick) were cut coronally with 80 µm intervals (total 400 µm, between -1.58 and -1.98 from bregma) [27] and mounted on gelatin-coated slides for cresyl violet staining, Fluoro-Jade staining, and glial cell immunostaining.
Cresyl violet and Fluoro-Jade staining
Cell death was evaluated using cresyl violet and Fluoro-Jade staining. Briefly, sections were serially hydrated using 100% ethanol to tap water. Next, they were incubated for 15 min in 0.1% cresyl violet solution. After destaining with 95% ethanol containing 0.1% glacial acetic acid, sections were dehydrated using a graded ethanol series (70% to 100%), followed by 100% xylene, and mounted with Canada balsam. Finally, cell death was evaluated under light microscopy (BX51, Olympus, Tokyo, Japan).
For Fluoro-Jade staining, sections were initially treated for 7 min at room temperature with 0.06% potassium permanganate. After washing with distilled water, sections were transferred to a solution of 0.001% Fluoro-Jade for 30 min with gentle shaking. Next, sections were dried for 1 h and dehydrated through a series of ethanol and 100% xylene. Finally, sections were mounted with DPX and observed using fluorescence microscopy (Axioimager M1, Carl Zeiss, Jena, Germany). Fluoro-Jade-positive cells in the CA1 and CA3 pyramidal cell layer of the hippocampus (shown in the white-colored boxes of Fig. 3) were counted.
Immunohistochemistry
Free-floating sections were washed 3 times (for 5 min each) with 0.01 M phosphate-buffered saline (PBS, pH 7.4). Sections were incubated for 15 min with 3% H2O2 and 10% methanol in 0.01 M PBS to destroy endogenous peroxidase activity. Next, sections were blocked for 1 h with 10% normal goat serum in 0.01 M PBS and incubated overnight at 4℃ with an antibody to GFAP (1:400 dilution) and Iba-1 (1:500 dilution). The following day, sections were incubated for 2 h at room temperature with anti-mouse IgG (1:500 dilution) or anti-rabbit IgG (1:200 dilution). Finally, sections were visualized with 0.1% DAB and 0.005% H2O2 in 0.05 M Tris HCl (pH 7.4) and observed using a light microscope (BX51, Olympus).
RESULTS
Influence of aspirin on seizure susceptibility
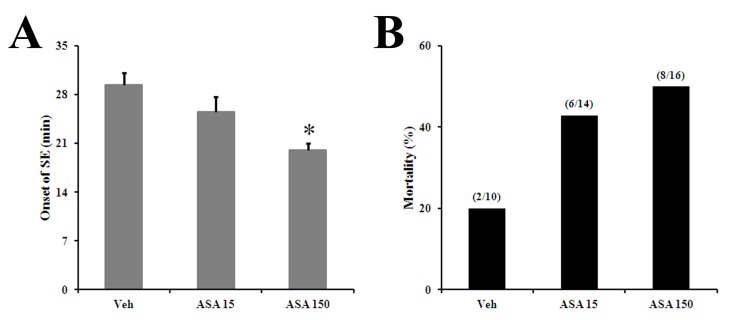
To investigate effect of aspirin on seizure susceptibility, onset time of SE, and mortality, mice were injected with pilocarpine as described in the Methods section. The onset of SE was significantly faster in the high dose (150 mg/kg) aspirin-treated group (ASA) compared with the vehicle-treated group (Veh) (Veh, 29.4±1.8 min; 15 mg/kg-ASA, 25.2±2.2 min; 150 mg/kg ASA, 20±1.0 min) (Fig. 1A). There was also increased mortality in the aspirin treated groups (Veh, 20%; 15 mg/kg-ASA, 42.9%; 150 mg/kg-ASA, 50%) (Fig. 1B).
Cresyl violet staining
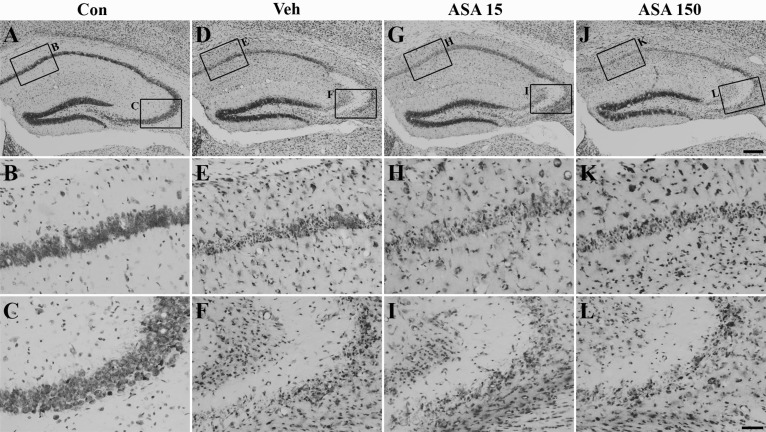
Neuronal cell death was evaluated with cresyl violet staining (Fig. 2). Viable cells were observed in the hippocampal CA1 and CA3 subfields of the normal control group (Fig. 2A~C), whereas many pyknotic nuclei were observed in the same regions of vehicle-treated mice 7 days after SE (Fig. 2D~F). In low (15 mg/kg) and high dose (150 mg/kg) aspirin-treated mice (Fig. 2G~I, 2J~L, respectively), neuronal cell death was not significantly different from vehicle-treated mice (Fig. 2D~F).
Fluoro-Jade staining and analyses
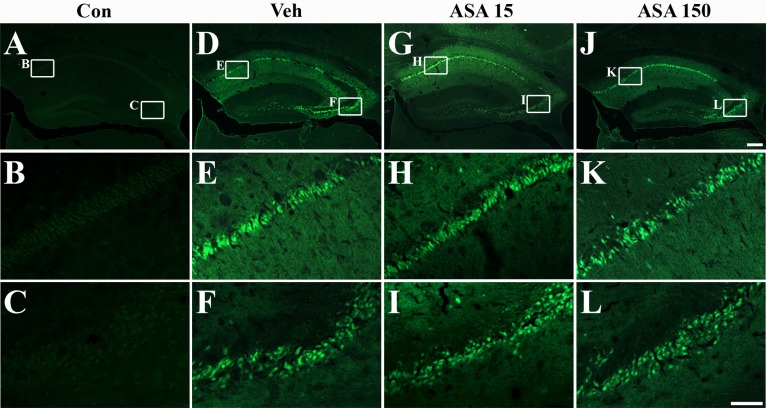
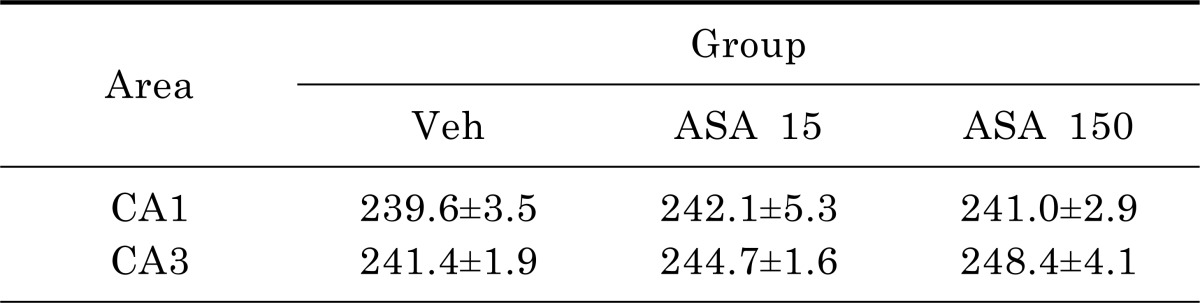
To quantitatively confirm the neuronal cell death, sections were labeled with Fluoro-Jade. Fluoro-Jade-positive cells were not detected in the hippocampus of normal mice (Fig. 3A~C). However, many dying or degenerating cells were observed in the hippocampal CA1 and CA3 pyramidal cell layers of vehicle (Fig. 3D~F), low-dose (Fig. 3G~I), and high-dose (Fig. 3J~L) aspirin-treated mice. However, quantitative analyses of Fluoro-Jade-positive cells did not reveal any significant differences between vehicle-, low dose, and high dose aspirin-treated mice (Table 1).
Reactive astrocytosis and activated microglia
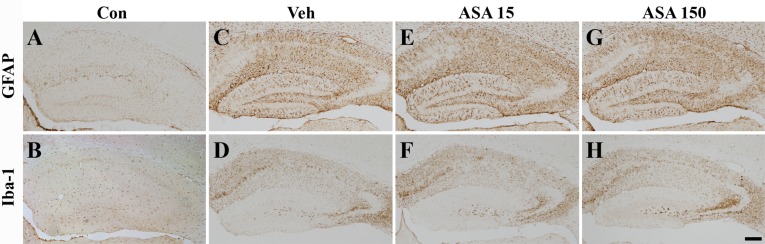
Neuronal cell death after SE evokes a considerable glial response in the hippocampus and glial responses are well-known markers of inflammation [28, 29]. Therefore, we examined the effects of aspirin on glial reaction by detecting GFAP and Iba-1 immunoreactivity (astrocyte and microglia markers, respectively) after pilocarpine-induced SE. GFAP- and Iba-1-immunoreactive cells were rarely observed in the hippocampus of normal mice (Fig. 4A, B). On the other hand, GFAP and Iba-1 immunoreactivity was increased throughout the hippocampus 7 days after SE (Fig. 4C, D). However, there was no distinct difference between low (Fig. 4E, F) and high (Fig. 4G, H) dose aspirin-treated and vehicle-treated animals.
DISCUSSION
Systemic administration of pilocarpine to rodents leads to epileptic injuries in the limbic system, which includes initial neuronal injuries, inflammation, marked gliosis, and hippocampal sclerosis. In the present study, we examined the effect of the anti-inflammatory agent aspirin on seizure susceptibility and neuropathology following pilocarpine-induced SE in mice. Our data suggest that aspirin treatment increases seizure susceptibility without a prominent effect on neuronal cell death and gliosis in the hippocampus.
In the present study, 15 mg/kg and 150 mg/kg of aspirin was administered daily for 10 days (3 days before to 6 days after SE). According to the interspecies scaling relationship formula from Mordentia and Chappell [25], 15 and 150 mg/kg of aspirin in mice is equivalent to 100 and 1,000 mg in a 70 kg-human, respectively. The low-dose of aspirin used in this study is much lower than the approved dose by the FDA to prevent thrombus formation (325 mg/kg per day). In addition, considering that the therapeutic dose of aspirin for pain and fever is 325~650 mg every 4~6 h, both the high and low doses of aspirin used in the present study are clinically applicable [30].
The main finding of this study is that aspirin administration increases seizure susceptibility and mortality. In agreement with our data, an active metabolite of aspirin, salicylate, increased seizure susceptibility in the pilocarpine-induced SE rat model [21]. In addition, treatment with sodium salicylate produced seizure in dogs [31] and increased susceptibility to acoustic trauma-induced audiogenic seizure in mice [32]. Furthermore, it has been reported that aspirin generated epileptic seizures in patients without a remarkable neurologic medical history [33]. Aspirin has also been reported to increase the expression of the N-methyl-D-aspartate receptor subunit 2A (NR2A) in the hippocampus [34] and it has been demonstrated that NMDA receptors play a key role in epileptogenesis [35, 36]. Blocking NR2A-containing NMDA receptors prevents pilocarpine-induced limbic seizure [37]. Furthermore, aspirin facilitates the Ca2+-dependent release of glutamate evoked by 4-aminopyridine [38]. It has been also suggested that salicylate acts as a non-competitive antagonist on glycine receptor containing α1 subunit [39]. Taken together, these data suggest that aspirin and/or its active metabolite, salicylate, could increase seizure susceptibility by directly activating excitatory neurotransmitter signaling systems and/or inhibiting inhibitory neurotransmitter signaling systems.
It is well known that pilocarpine-induced SE induces neuronal cell death, followed by a marked glial response in several brain regions, including the hippocampus. In this study, neuronal cell death and glial activation in the hippocampus increased 7 days after pilocarpine-induced SE. Interestingly, we did not observe any significant differences in neuronal cell death and glial response between vehicle- and aspirin-treated mice following SE. This result is supported by a previous study showing that SC-58236, a COX-2 inhibitor, significantly increased mortality and the spontaneous seizure tendency, without significant effects on cell death, microglial activation, and mossy fiber sprouting [40]. However, it has been reported that administration of sodium salicylate exacerbated neuronal cell death in the hippocampal CA1, dentate gyrus, dorsomedial thalamic nucleus, and entorhinal cortex and increased microglial activation in kainic acid-induced seizure [22]. In addition, COX-2 selective inhibitors, such as NS-398 and celecoxib, and a nonselective COX inhibitor, indomethacin, were reported to reduce seizure threshold and exacerbate neuronal cell damage in a kainic acid-induced SE model [41]. On the other hand, Ma et al. recently delineated that aspirin administration decreased neuronal cell death in the CA1, CA3, and hilus of the hippocampus in a rat model of lithium-pilocarpine-induced SE and attenuated mossy fiber sprouting and spontaneous recurrent seizure after SE [23]. Several reports have provided evidence supporting the beneficial effects of COX inhibitors against epilepsy in rodents [42,43]. Taken together, the data indicate that the effect of anti-inflammatory agents, including aspirin, on neuropathology induced by SE is controversial and these conflicting results may be due to the differences in experimental conditions, such as the seizure type, drug schedule, and experimental protocol. Further studies are needed to clarify the effect of NSAIDs including aspirin on neuronal damage and glial responses after epileptic seizures.
In summary, aspirin significantly increased seizure susceptibility and mortality following pilocarpine-induced SE without affecting neuronal cell death and glial response in the mouse hippocampus. The present study provides further evidence that the therapeutic use of aspirin in the patients with neurological disorders such as TLE requires reevaluation.
 XML Download
XML Download