

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Functional defects in mitochondria are involved in the induction of cell death in cancer cells. We assessed the toxic effect of camptothecin against the human cervical and uterine tumor cell line SiHa with respect to the mitochondria-mediated cell death process, and examined the combined effect of camptothecin and anticancer drugs. Camptothecin caused apoptosis in SiHa cells by inducing mitochondrial membrane permeability changes that lead to the loss of mitochondrial membrane potential, decreased Bcl-2 levels, cytochrome c release, caspase-3 activation, formation of reactive oxygen species and depletion of GSH. Combination of camptothecin with other anticancer drugs (carboplatin, paclitaxel, doxorubicin and mitomycin c) or signaling inhibitors (farnesyltransferase inhibitor and ERK inhibitor) did not enhance the camptothecin-induced cell death and caspase-3 activation. These results suggest that camptothecin may cause cell death in SiHa cells by inducing changes in mitochondrial membrane permeability, which leads to cytochrome c release and activation of caspase-3. This effect is also associated with increased formation of reactive oxygen species and depletion of GSH. Combination with other anticancer drugs (or signaling inhibitors) does not appear to increase the anti-tumor effect of camptothecin against SiHa cells, but rather may reduce it. Combination of camptothecin with other anticancer drugs does not seem to provide a benefit in the treatment of cervical and uterine cancer compared with camptothecin monotherapy.
REFERENCES
Ackermann S., Beckmann MW., Thiel F., Bogenrieder T. Topotecan in cervical cancer. Int J Gynecol Cancer. 17:1215–1223. 2007.

Arbuck SG., Takimoto CH. An overview of topoisomerase I-targeting agents. Semin Hematol. 35(Suppl 4):3–12. 1998.
Armstrong JS. Mitochondria: a target for cancer therapy. Br J Pharmacol. 147:239–248. 2006.
Berthier A., Lemaire-Ewing S., Prunet C., Monier S., Athias A., Bessede G., Pais de Barros JP., Laubriet A., Gambert P., Nèel D. Involvement of a calcium-dependent dephosphorylation of BAD associated with the localization of Trpc-1 within lipid rafts in 7-ketocholesterol-induced THP-1 cell apoptosis. Cell Death Differ. 11:97–905. 2004.
Blum R., Jacob-Hirsch J., Rechavi G., Kloog Y. Suppression of survivin expression in glioblastoma cells by the Ras inhibitor farnesylthiosalicylic acid promotes caspase-dependent apoptosis. Mol Cancer Ther. 5:2337–2347. 2006.
Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1411:351–369. 1999.
Chandra J., Samali A., Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 29:323–333. 2000.
Chang F., Steelman LS., Shelton JG., Lee JT., Navolanic PM., Blalock WL., Franklin R., McCubrey JA. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway. Int J Oncol. 22:469–480. 2003.
Constantini PC., Chernyak BC., Petronilli V., Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem. 271:6746–6751. 1996.
Crow MT., Mani K., Nam YJ., Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 95:957–970. 2004.
Dias N., Bailly C. Drugs targeting mitochondrial functions to control tumor cell growth. Biochem Pharmacol. 70:1–12. 2005.
Faivre S., Djelloul S., Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol. 33:407–420. 2006.
Ferreira CG., Span SW., Peters GJ., Kruyt FA., Giaccone G. Chemotherapy triggers apoptosis in a caspase-8-dependent and mitochondria-controlled manner in the non-small cell lung cancer cell line NCI-H460. Cancer Res. 60:7133–7141. 2000.
Fleury C., Mignotte B., Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 84:131–141. 2002.
Fu W., Luo H., Parthasarathy S., Mattson MP. Catecholamines potentiate amyloid β-peptide neurotoxicity: involvement of oxidative stress, mitochondrial dysfunction, and perturbed calcium homeostasis. Neurobiol Dis. 5:229–243. 1998.
Hall AG. The role of glutathione in the regulation of apoptosis. Eur J Clin Invest. 29:238–245. 1999.
Hong JS., Ko HH., Han ES., Lee CS. Inhibition of bleomycin-induced cell death in rat alveolar macrophages and human lung epithelial cells by ambroxol. Biochem Pharmacol. 66:1297–1306. 2003.
Huang X., Kurose A., Tanaka T., Traganos F., Dai W., Darzynkiewicz Z. Activation of ATM and histone H2AX phosphorylation induced by mitoxantrone but not by topotecan is prevented by the antioxidant N-acetyl-L-cysteine. Cancer Biol Ther. 5:959–964. 2006.
Kim R., Emi M., Tanabe K. Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol. 57:545–553. 2006.
Kobayashi T., Sawa H., Morikawa J., Jhang W., Shiku H. Bax induction activates apoptotic cascade via mitochondrial cytochrome c release and Bax overexpression enhances apoptosis induced by chemotherapeutic agents in DLD-1 colon cancer cells. Jpn J Cancer Res. 91:1264–1268. 2000.
Lee CS., Kim YJ., Lee MS., Han ES., Lee SJ. 18β-Glycyrrhetinic acid induces apoptotic cell death in SiHa cells and exhibits a synergistic effect against antibiotic anti-cancer drug toxicity. Life Sci. 83:481–489. 2008.
Lee CS., Park SY., Ko HH., Han ES. Effect of change in cellular GSH levels on mitochondrial damage and cell viability loss due to mitomycin c in small cell lung cancer cells. Biochem Pharmacol. 68:1857–1867. 2004.
Mignotte B., Vayssière JL. Mitochondria and apoptosis. Eur J Biochem. 252:1–15. 1998.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 65:55–63. 1983.
Nitiss JL., Wang JC. Mechanisms of cell killing by drugs that trap covalent complexes between DNA topoisomerases and DNA. Mol Pharmacol. 50:1095–1102. 1996.
Pirnia F., Schneider E., Betticher DC., Borner M. Mitomycin c induces apoptosis and caspase-8 and −9 processing through a caspase-3 and Fas-independent pathway. Cell Death Differ. 9:905–914. 2002.
Porter SE., Champoux JJ. The basis for camptothecin enhancement of DNA breakage by eukaryotic topoisomerase I. Nucleic Acids Res. 17:8521–8532. 1989.
Pritsos CA., Briggs LA., Gustafson DL. A new cellular target for mitomycin c: a case for mitochondrial DNA. Oncol Res. 9:333–337. 1997.
Sane AT., Cantin AM., Paquette B., Wagner JR. Ascorbate modulation of H2O2 and camptothecin-induced cell death in Jurkat cells. Cancer Chemother Pharmacol. 54:315–321. 2004.
Shao RG., Cao CX., Zhang H., Kohn KW., Wold MS., Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA: DNA-PK complexes. EMBO J. 18:1397–1406. 1999.
Timur M., Akbas SH., Ozben T. The effect of Topotecan on oxidative stress in MCF-7 human breast cancer cell line. Acta Biochim Pol. 52:897–902. 2005.
Tsao YP., Russo A., Nyamuswa G., Silber R., Liu LF. Interaction between replication forks and topoisomerase I-DNA cleavable complexes: studies in a cell-free SV40 DNA replication system. Cancer Res. 53:5908–5914. 1993.
Van Klaveren RJ., Hoet PHM., Pype JL., Demedts M., Nemery B. Increase in γ-glutamyltransferase by glutathione depletion in rat type II pneumocytes. Free Radic Biol Med. 22:525–534. 1997.
Wang P., Song JH., Song DK., Zhang J., Hao C. Role of death receptor and mitochondrial pathways in conventional chemotherapy drug induction of apoptosis. Cell Signal. 18:1528–1535. 2006.
Wenzel U., Nickel A., Kuntz S., Daniel H. Ascorbic acid suppresses drug-induced apoptosis in human colon cancer cells by scavenging mitochondrial superoxide anions. Carcinogenesis. 25:703–712. 2004.
Xia S., Rosen EM., Laterra J. Sensitization of glioma cells to Fas-dependent apoptosis by chemotherapy-induced oxidative stress. Cancer Res. 65:5248–5255. 2005.
Yang X., Zhang C., Ying M., Yang B., He Q. Antiproliferation in human EA.hy926 endothelial cells and inhibition of VEGF expression in PC-3 cells by topotecan. Pharmazie. 62:534–538. 2007.
Fig. 1.
Camptothecin induces cell viability loss. SiHa cells were treated with either 0.5~5 μM camptothecin (CPT) for 24 h and then cell viability was determined by using the MTT assay. Data represent mean±SEM (n=6). p<0.05 compared to control (percentage of control).
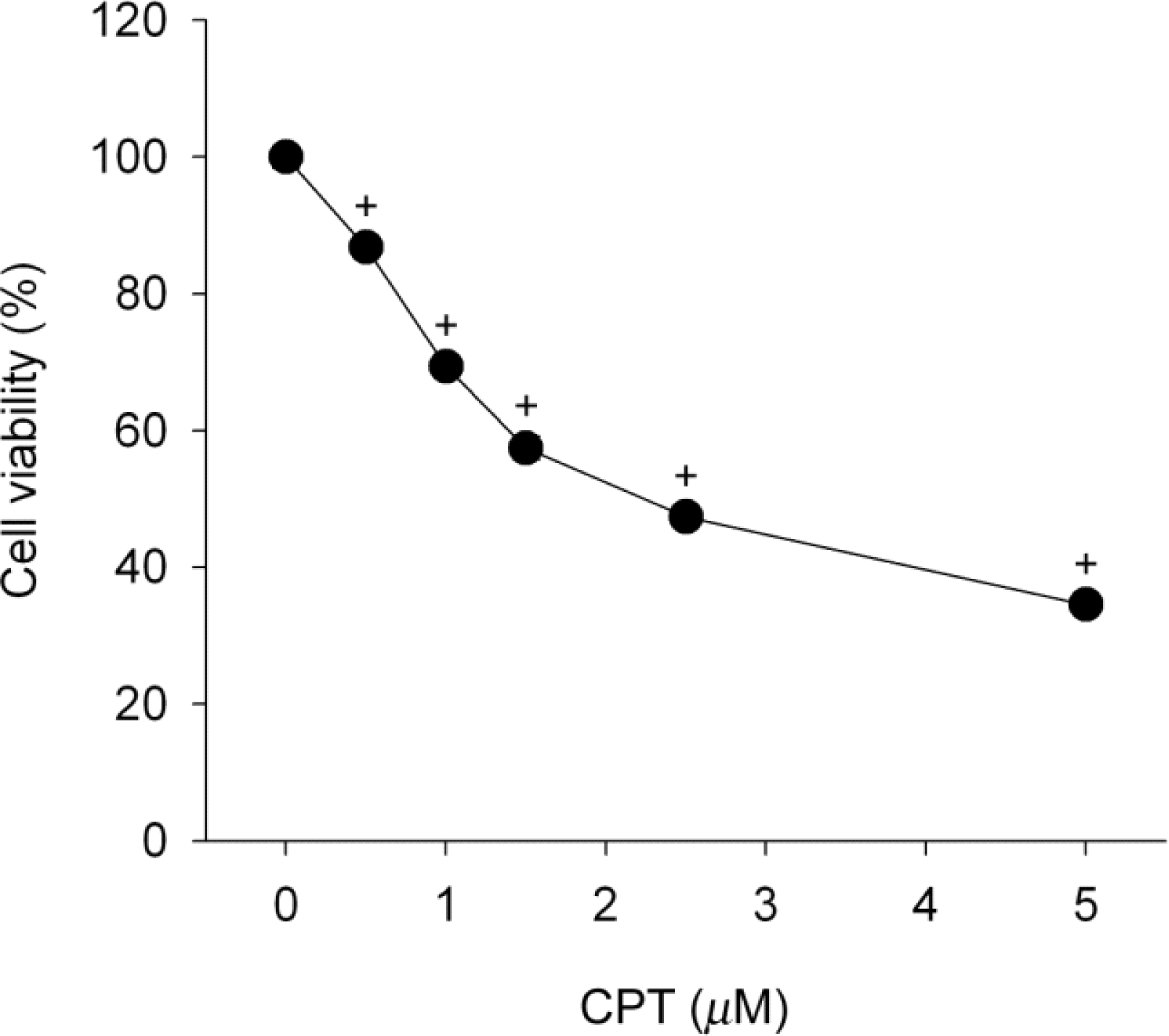
Fig. 2.
Camptothecin induces cytochrome c release and caspase-3 activation. SiHa cells were treated with 1~5 μM camptothecin (CPT) for 24 h. In the assay (A) for the mitochondrial transmembrane potential, data are expressed as the percentage of cells with depolarized mitochondria for the mitochondrial membrane potential and represent mean±SEM (n=4). In western blot assay (B), the levels of Bcl-2 and cytochrome c in the cytosolic fractions were analyzed by western blotting with antibodies (anti Bcl-2 and anti-cytochrome c). Data are representative of three different experiments. In ELISA-based quantitative analysis (C), values represent ng/ml for cytochrome c release and arbitrary units (a.u.) for caspase-3 activity. Data are mean±SEM (n=6). p<0.05 compared to control.
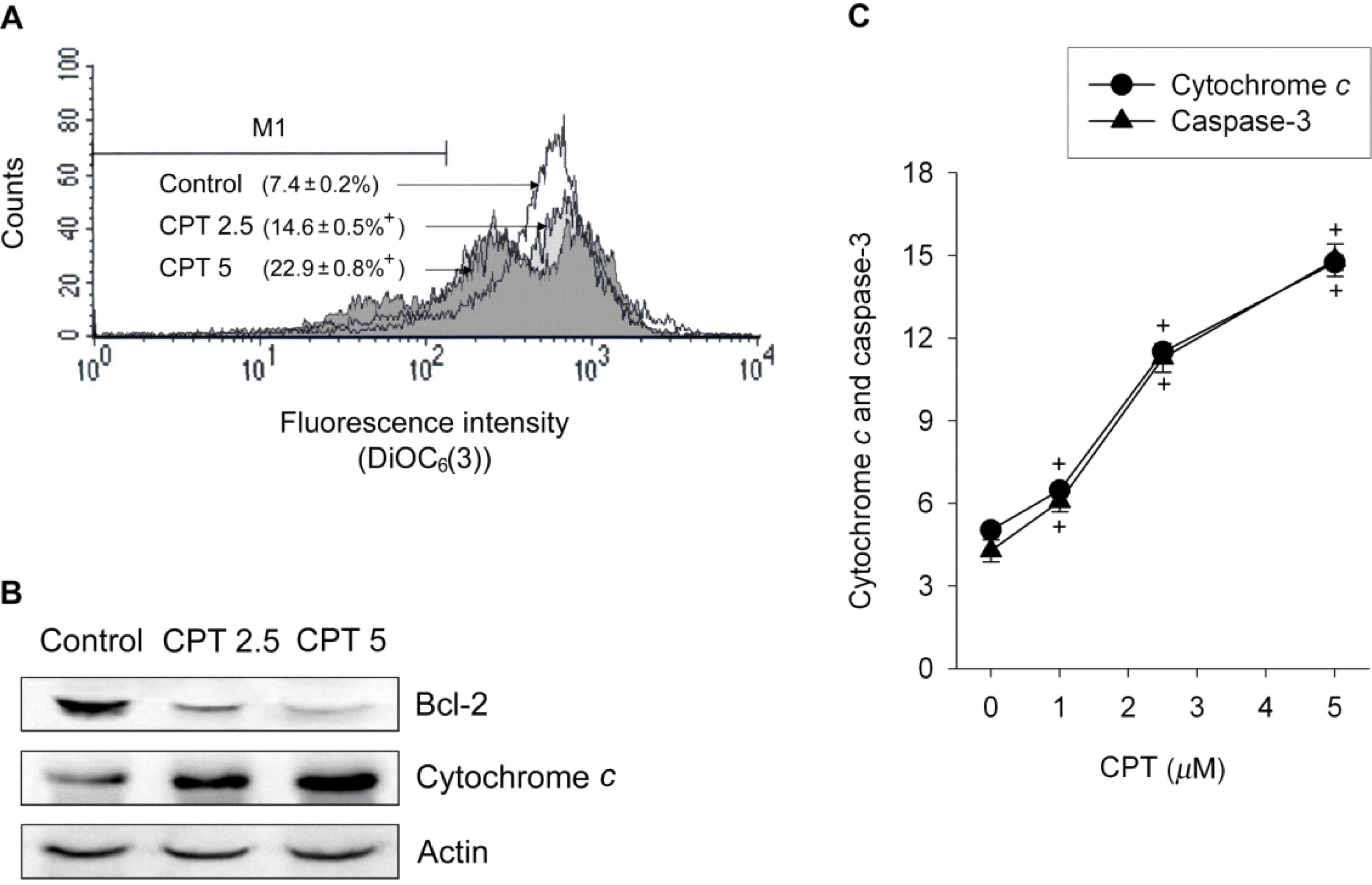
Fig. 3.
Camptothecin induces formation of reactive oxygen species. In experiment (A) for reactive oxygen species formation, SiHa cells were treated with either 1 ~ 5 μM camptothecin (CPT) or 2.5 μM camptothecin plus 1 mM N-acetylcysteine (NAC) for 24 h. Data are expressed as arbitrary units of fluorescence (a.u.) and represent mean±SEM (n=6). p<0.05 compared to control; and ∗p<0.05 compared to camptothecin alone. In experiment (B) for cell death, SiHa cells were treated with 2.5 μM camptothecin (CPT) in combination with scavengers [(1 mM N-acetylcysteine (NAC), 30 μM trolox, 30 μM carboxy-PTIO (PTIO) and 1 mM ascorbate (Asc)] for 24 h, and cell viability was determined. Data represent mean±SEM (n=6). p<0.05 compared to control; and ∗p<0.05 compared to camptothecin alone.
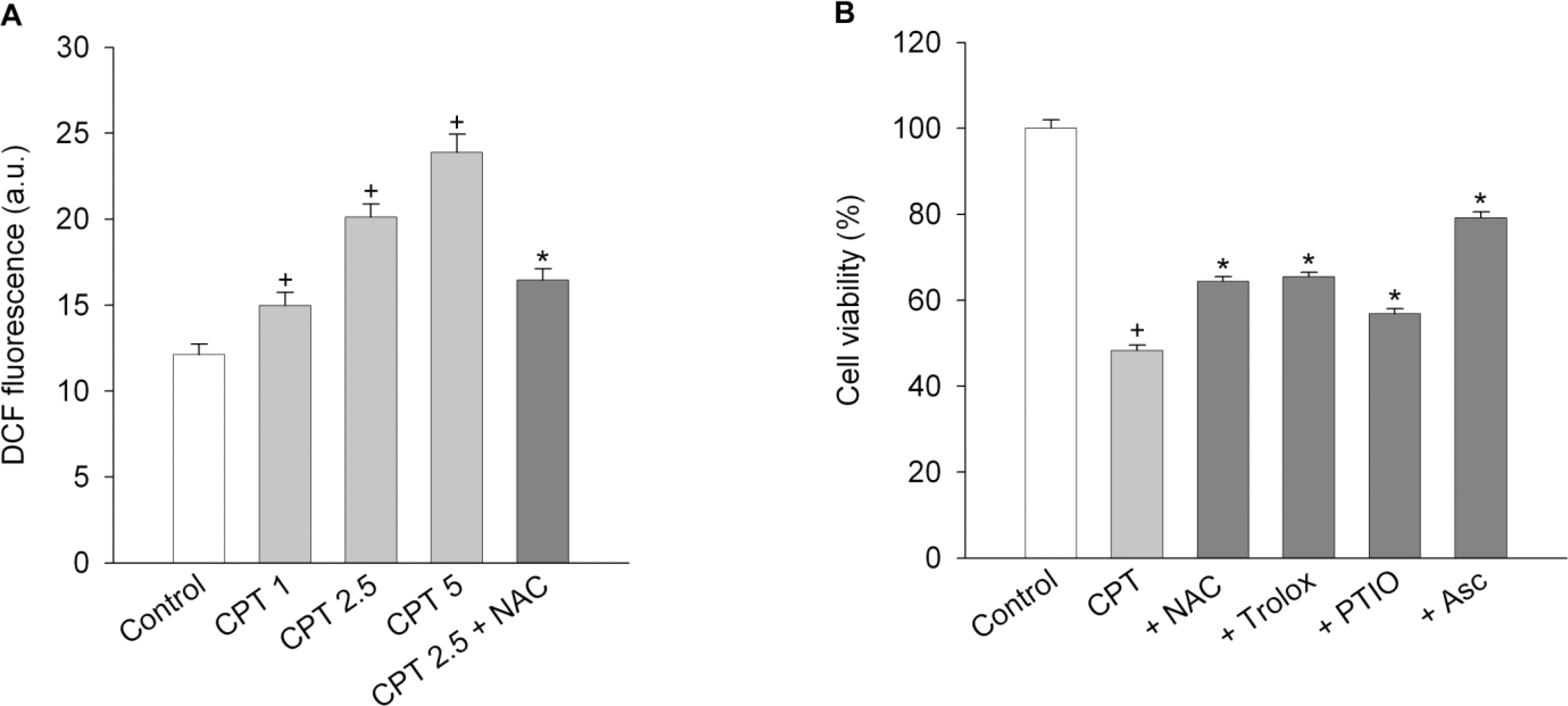
Fig. 4.
Camptothecin induces depletion of GSH contents. SiHa cells were treated with 0.5~5 μM camptothecin (CPT) for 24 h. Data are expressed as nmol of GSH/mg protein and represent mean±SEM (n=6). +p<0.05 compared to control.
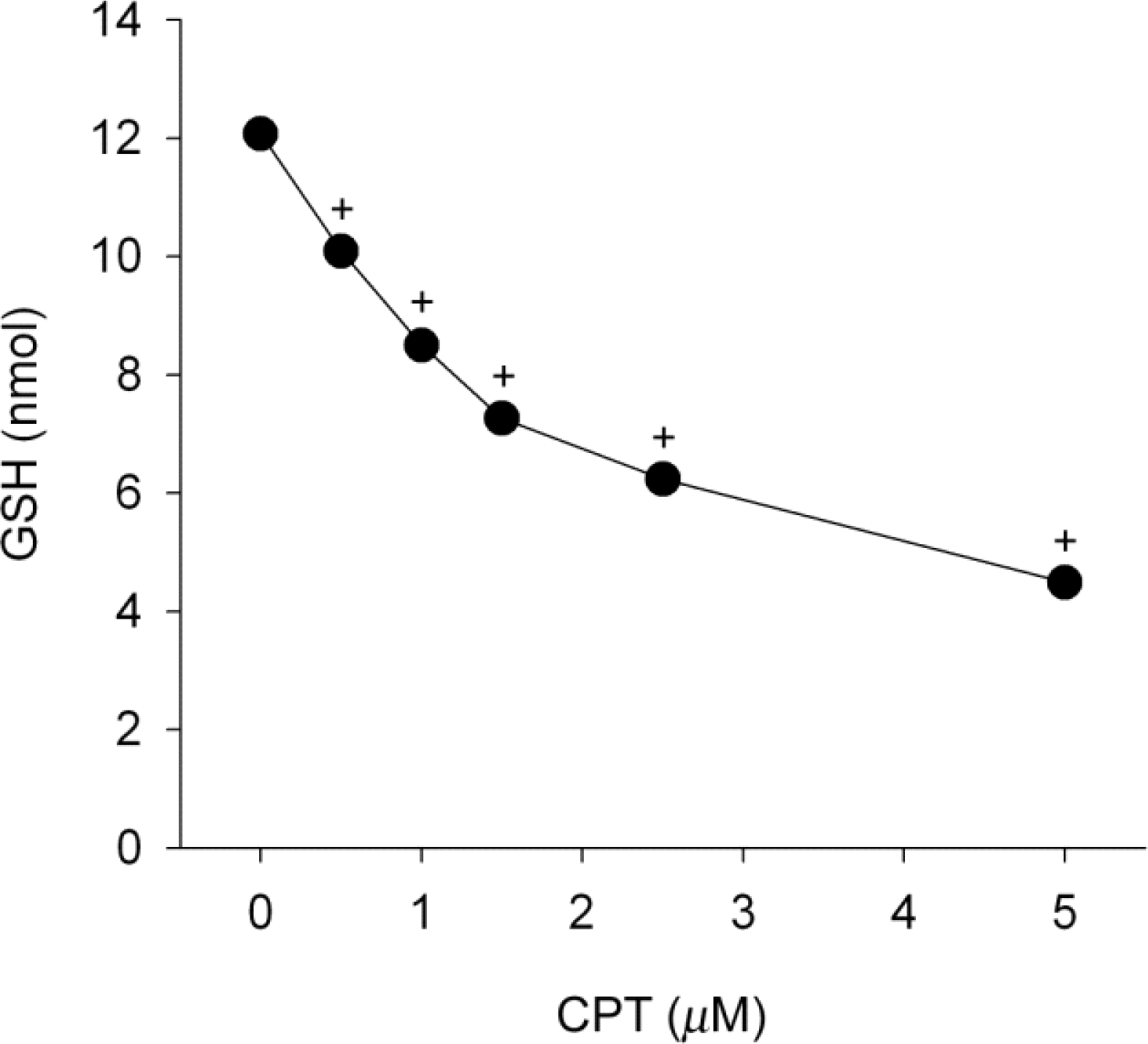
Fig. 5.
Effect of anticancer drugs on camptothecin-induced cell death. SiHa cells were treated with 2.5 μM camptothecin (CPT) in combination with anticancer drugs [100 μM carboplatin (Carbo), 100 μM paclitaxel (Pacli), 10 μM doxorubicin (Doxo) or 3 μg/ml mitomycin c (MMC)] for 24 h, and then cell viability was measured by using MTT reduction assay (A) or neutral red uptake assay (B). Data represent mean±SEM (n=6). +p<0.05 compared to control; and ∗p<0.05 compared to camptothecin alone.
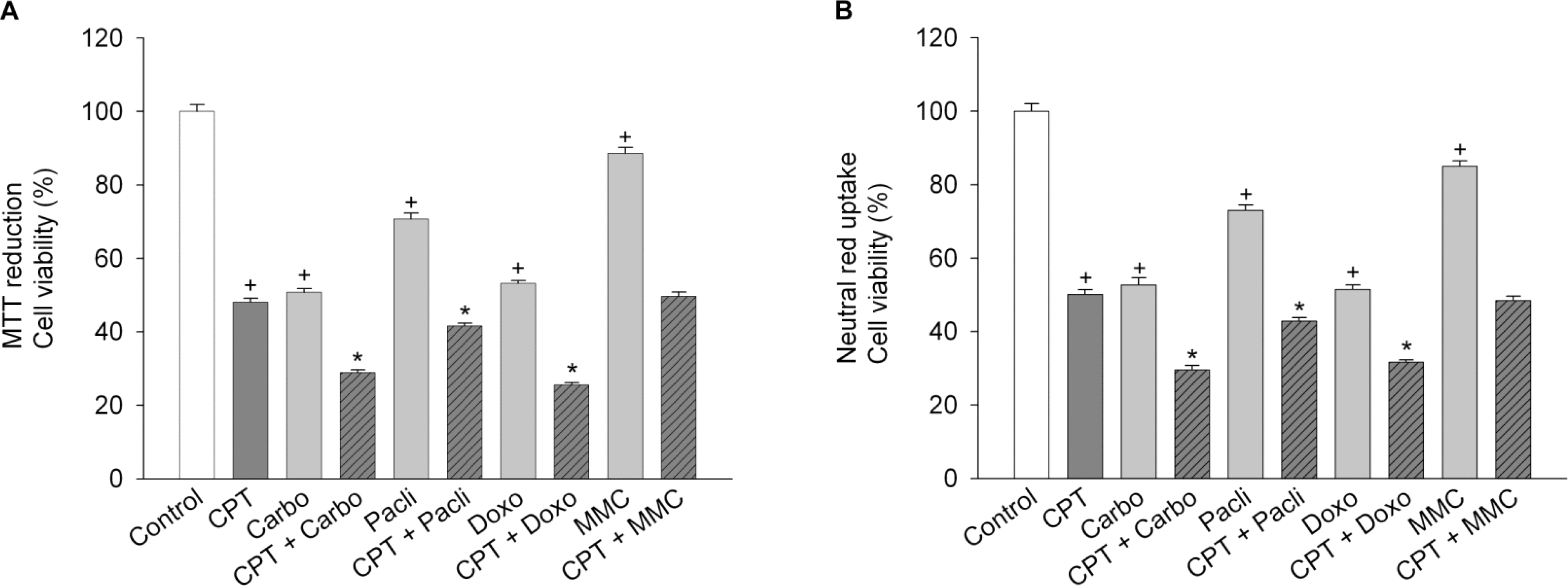
Fig. 6.
Effect of signaling inhibitors on camptothecin-induced cell death. SiHa cells were treated with 2.5 μM camptothecin (CPT) in combination with 0.5 μM FTI [or 5 μM ERK inhibitor (ERKi)] for 24 h and then cell viability was measured. Data represent mean±SEM (n=6). +p<0.05 compared to control; and ∗p<0.05 compared to camptothecin alone.
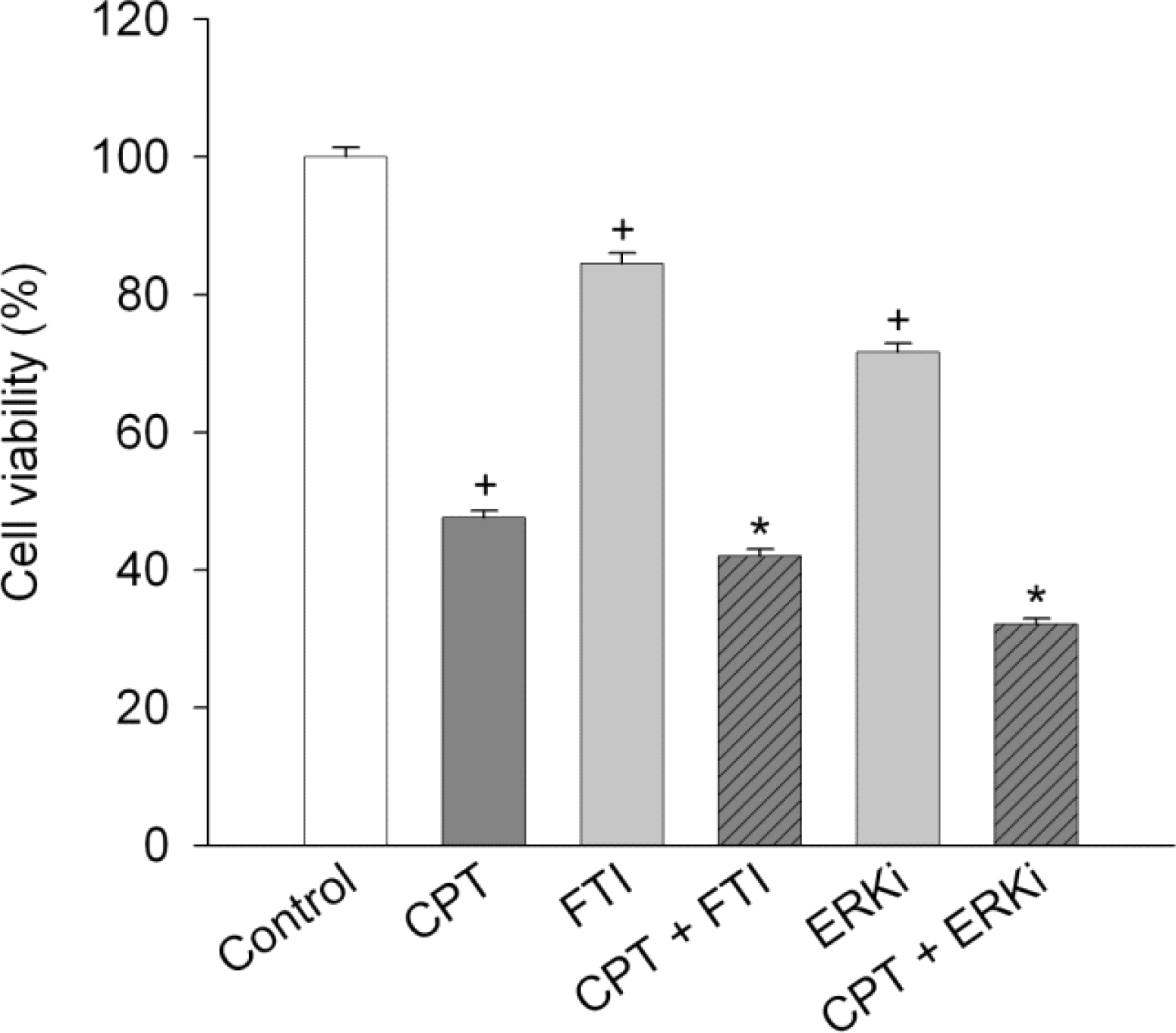
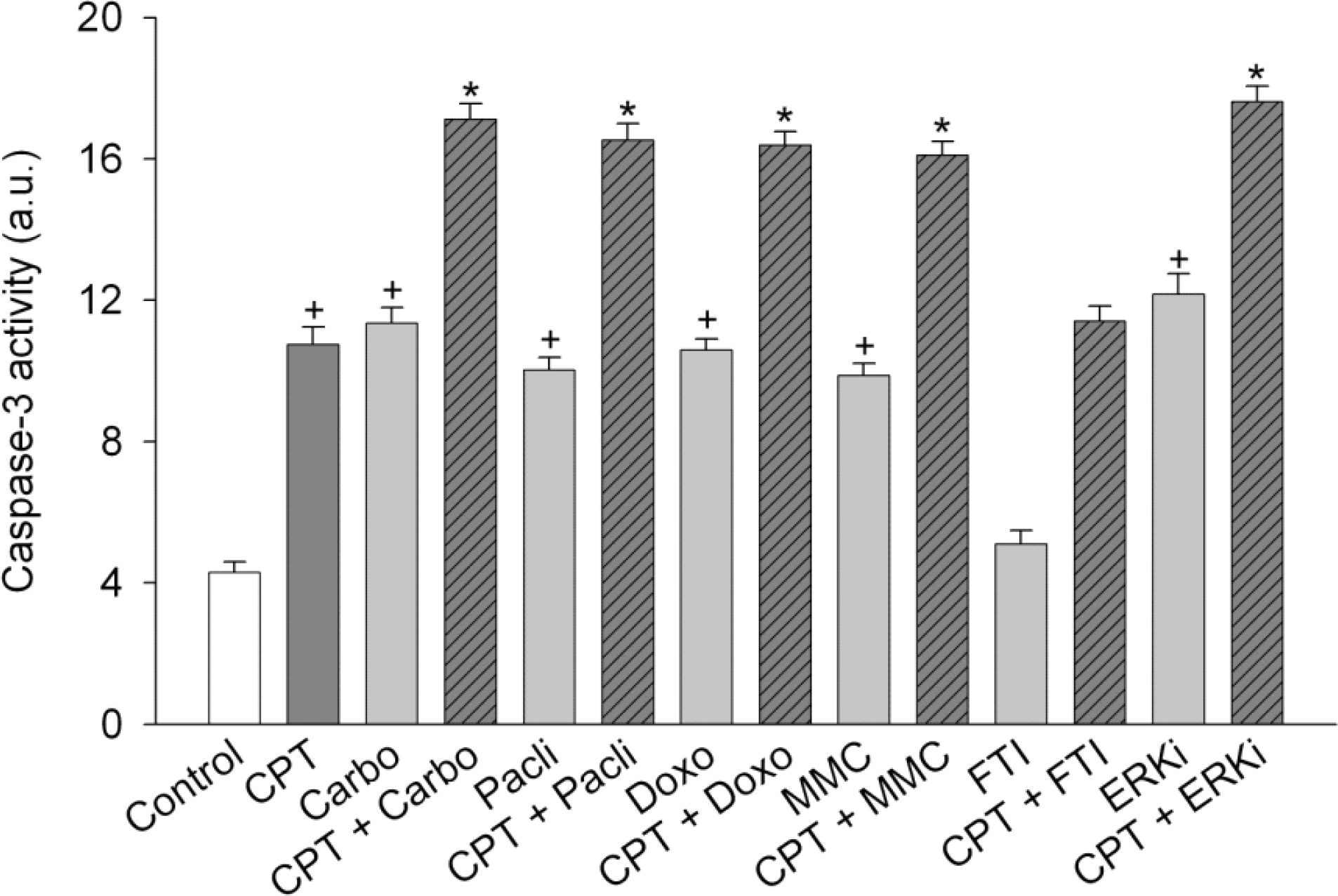
Fig. 7.
Effect of anticancer drugs on camptothecin-induced activation of caspase-3. SiHa cells were treated with 2.5 μM camptothecin (CPT) in combination with anticancer drugs [100 μM carboplatin (Carbo), 100 μM paclitaxel (Pacli), 10 μM doxorubicin (Doxo) or 3 μg/ml mitomycin c (MMC)] or signaling inhibitors [0.5 μM and 5 μM ERK inhibitor (ERKi)] for 24 h. Data are expressed as units for caspase-3 activity and represent mean±SEM (n=6). +p <0.05 compared to control; and ∗p<0.05 compared to camptothecin alone.
 XML Download
XML Download