

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Familial Mediterranean fever (FMF) is an autosomal recessive disease characterized by the manifestation of symptoms such as recurrent episodes of fever with serosal, synovial, or cutaneous inflammation (1). Because of its genetic characteristic, this disease is found in around 0.1%-0.3% of high-risk population groups including Turkish, Armenian, Arabian, and Jews in the Mediterranean and the Middle East. Since the mutation of Mediterranean fever gene (MEFV) was found to play a major role in the onset of the disease (2), the significant numbers of FMF have been found in North african countries, Greece, Crete, France, Germany, Italy, US, and even Japan. In general, the onset age is 20 yr or younger in 90% of the patients and half of them have the symptoms before 10 (3). Nevertheless, the one and only case of FMF in Korea was the patient diagnosed before 10 (4). Onset of the disease at an older age may occur but is rare (5). We report the case of adult-onset FMF in Korea who presented with recurrent periodic fever, abdominal pain and vomiting.
CASE DESCRIPTION
A 32-yr-old male patient visited infection clinic of our hospital for fever, abdominal pain and vomiting in April 2011. He had no past medical history, family history and history of tobacco, alcohol, illegal drug use, and travel. He had been suffering from recurrent periodic fever, abdominal pain, and vomiting every three months since 3 yr ago. He complained of abdominal discomfort for two days and fever for three days followed along with whole abdominal pain and vomiting, and then the symptoms resolved spontaneously and completely. He could maintain a normal daily life during the symptom free period. He had already taken several blood tests, abdominal CT, gastroscopy, and colonoscopy a year ago in two another hospitals, but the cause of his symptoms had not been found.
His vital signs were blood pressure 120/68 mmHg, pulse 97/min, respiratory rate 22/min, and body temperature 40.0℃ on admission. Mild tenderness and hypoactive bowel sound were checked on the whole abdomen. Initial laboratory results showed that leukocyte was 9,980/µL (neutrophil 8,830/µL), hemoglobin 16.8 g/dL, platelet 171,000/µL, C-reactive protein 85.71 mg/L, and the erythrocyte sedimentation rate 6 mm/hr. Various antibody tests screening for infection, antibody tests for autoimmune disease, blood complement levels, blood cultures, peripheral blood smear, tumor markers, and hormone tests were all negative.
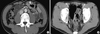
In abdominal CT with contrast enhancement (Fig. 1), were observed edematous wall thickening of proximal jejunum, dilatation of proximal jejunum, accompanied by lymphadenopathy, and a small amount of ascites. Therefore capsule endoscopy was done for the evaluation of small intestine. Mild lymphangiectasia was observed throughout the small intestine in capsule endoscopy. Despite biopsies taken at suspicious lymphangiectasia through double balloon enterosocpy, there was non-specific, only mild inflammation of mucosa. His symptoms lasted for around 3 days and then were spontaneously subsided, and abdominal ultrasonography on the 4th day of admission showed that jejunal dilatation and lymphadenopathy almost completely disappeared.
Through several tests, we could exclude several suspected diseases such as infectious diseases, autoimmune diseases and neoplastic diseases, and based on his repetitive symptoms and age, we thought that it was highly likely to be FMF or tumor necrosis factor receptor-associated periodic syndrome (TRAPS) among periodic fever syndromes. We performed MEFV and TNFR1 gene test for definite diagnosis, and as two missense mutations were detected in the exon 2 of the MEFV gene, c.329T > C (p.Leu110Pro) in heterozygous and c.442G > C (p.Glu148Gln) in homozygous, this patient was diagnosed as FMF (Fig. 2, Table 1).
The patient is current under treatment with colchicine 1.2 mg/day and the progress is being followed up with gradually improved severity and duration of the symptoms.
DISCUSSION
FMF is a genetic disease in which the mutation of the MEFV gene changes the function of protein playing the role of inhibiting inflammation and, consequently, causes autoinflammation. And it was known to be that prevalent among Mediterraanean ethnic group. However, since the development of gene testing, the disease has been reported in unexpected and non-Mediterranean area, like Japan (6).
A major symptom of this disease is recurrent periodic fever accompanied with pain in abdomen, chest or joint. Abdominal pain is most common, observed in around 90% of the patients, and chest pain is observed in 20%-40% and joint pain in 50%-60%. In general, an important feature of the disease is the young age of onset; most patients (90%) begin to suffer before 20 yr of age, and 60% are younger than 10. Onset of the disease at an older age (after 40) may occur but is rare (5). The incidence of FMF with disease onset after 20 yr of age (adult-onset) is 14% and after 40 yr of age (late-onset) is only 0.5% in Turkey (5, 7). The symptoms last for 1-4 days and after recovery from the symptoms the patient can lead a normal life. Sometimes if the symptoms are severe the case is misdiagnosed as appendicitis and receives unnecessary abdominal surgery, and if the symptoms are mild it may be misdiagnosed as irritable bowel syndrome. This patient also had been thoroughly examined in two another hospitals, but could not find the cause of the symptoms. In case the disease infiltrates into a joint, it is usually manifested as monoarthritis and is sometimes accompanied with rash, and may be misdiagnosed as acute rheumatic fever, juvenile rheumatoid arthritis. FMF patients may have accompanying vasculitis such as Henoch-Schonlein purpura and polyarteritis nodosa, and if not treated properly, they may have progressive systemic amyloidosis that causes renal failure and even death (8, 9). Adult-onset FMF may be a form of disease with distinct clinical, demographic and molecular characteristics. Late-onset FMF emerged as a mild disease presenting with abdominal attacks and usually lacking other types of acute attacks and chronic or protracted manifestations, characterized by favorable response even to low dose colchicines (7).
FMF can be diagnosed based on characteristic clinical patterns, response to treatment for colchicines, and gene test results. In the endemic areas of FMF, provisional diagnosis is made through clinical pattern and family history, and definite diagnosis is made through response to colchicine treatment. But in non-endemic areas like our patient, it can be diagnosed through verifying the mutation of the MEFV gene (10).
The MEFV gene, which was thought as the cause of FMF, was discovered first in 1997. It is located on the short arm of chromosome 16. This gene is related to the coding of protein called pyrin (or marenostrin), which is manifested in neutrophil and plays the role of inhibiting inflammatory response, and it is known that if this protein is regulated abnormally due to gene mutation, neutrophil is activated excessively in serosa or elsewhere, and causes the acute onset of FMF (11).
Our patient had heterozygous for the p.Leu110Pro mutation located in exon 2. This is a previously reported mutation, and to date, several FMF patients have been reported to be compound heterozygote with other mutations (12). In addition, he also had homozygous for the p.Glu148Gln mutation located in exon 2. The carrier frequency of the p.Glu148Gln mutation has been reported to be 12% in Turks, 10% in Ashkenazi Jews, 6.4% in Jews of Moroccan origin, and as high as 53% in Jews from the various ethnic groups (13). It remains controversial whether the p.Glu148Gln mutation is a disease-causing mutation or a simple polymorphism because of high allele frequency in healthy controls. However, it has been reported that most homozygote or compound heterozygote patients associated with other MEFV mutations are symptomatic (14). In summary of the above results, this patient carried p.Leu110Pro and p.Glu148Gln mutations on an allele and carried p.Glu148Gln mutation on the other allele. One study was conducted in Japan that described these types of mutations have been reported in 5% of FMF patients in Japan (14).
Currently as of August, 2012, 227 gene mutations have been found and most of them are missense mutations. Among them, 100 gene mutations have been found to be associated with the manifestation of diseases (An updated list of mutations for FMF can be found online at http://fmf.igh.cnrs.fr/infevers/).
The goals of FMF treatment are to prevent the acute onset, avoid unnecessary development into amyloidosis, and relatively these goals are achieved successfully by medication with colchicine. According to a study on the long-term efficacy of colchicines conducted in 1991, 87% of 45 patients who took colchicines at a preventive dose of 1.0-3.0 mg/day for 15 yr responded to the treatment and the responses were decreases in all of the frequency, severity, and period of symptoms (15). In case patients do not respond to oral administration of colchicines, first of all, the patient's compliance should be evaluated and then the patient deserves further evaluation. There are several therapeutic options including interferon α, thalidomide, etanercept, and infliximab for the patients resistant to colchicines (16-19).
It is a limitation that we could not confirm the inheritance pattern of the two mutant alleles due to the lack of family study data. In this case, there were typical repetitive symptoms, and other diseases were excluded such as infectious disease, autoimmune disease, and neoplastic diseases through several tests, that we could finally diagnose the patient as FMF through MEFV gene test. In addition, his symptoms disappeared by taking colchicine.
We hope this case will raise the awareness of physicians that FMF should be considered in patients with periodic fever and abdominal pain in non-Mediterranean regions like Korea.


 XML Download
XML Download