

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pancreatic endocrine tumors are uncommon, with an annual incidence of 1-2 cases/100,000, but their frequency is on the rise (1). In younger individuals, hereditary cancer syndromes such as multiple endocrine neoplasia type 1 (MEN-1) may contribute to these cases. Pancreatic endocrine tumors are classified as functioning or non-functioning based on the presence or absence of a clinical symptom. Endocrine tumors of the pancreas are a heterogeneous group of neoplasms with natural histories varying from a frequently indolent course for tumors that are well differentiated to a much more aggressive form for poorly differentiated tumors (2). Due to their heterogeneity and rarity, improving the management of these tumors is particularly difficult. In general, surgical resection is the procedure of choice for patients with localized disease or limited metastases (3). In patients with advanced disease, clinical trials of immunotherapy and cytotoxic chemotherapy have demonstrated the efficacy of several drugs, including interferon alpha, somatostatin, streptozocin, doxorubicin, fluorouracil, dacarbazine, and temozolomide (4).
To date, however, systemic therapies for endocrine tumors have been largely ineffective. Although treatment with somatostatin analogs can usually decrease hormonal symptoms, it rarely results in tumor shrinkage. Therefore, although pancreatic endocrine tumors are indolent, the prognosis of patients with advanced tumors is poor, particularly when the condition progresses to a stage at which cytotoxic agents are not effective. Presently, drugs targeted to particular molecules involved in endocrine tumors are in clinical trials. Herein, we report on a patient with advanced pancreatic endocrine tumors and liver metastasis who responded favorably to sorafenib after the failure of biotherapy and conventional chemotherapy.
CASE DESCRIPTION
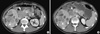
A 31-yr-old man visited our hospital following 10 days of abdominal pain on February 14, 2006. His past history was unremarkable, as was his family history. His initial computed tomography (CT) and positron emission tomography scans showed a 6.8 cm lobulated mass in the uncinate process of the pancreas without invasion of the adjacent vessels and the common bile duct. The tumor showed focal cystic and necrotic areas, and heterogeneous enhancement. Multiple hematogenous metastases were also noted in the liver (Fig. 1A). Percutaneous needle biopsy of a hepatic nodule was performed to establish histological diagnosis. Atypical cells with an acinar formation were noted but their histological origin was unknown. Multiple hepatic segmentectomy and pancreatic mass resection were performed to confirm the histological diagnosis. The tumor cells were arranged in trabeculae and solid nests separated by a fibrous or loose fibrovascular stroma. All tumor cells were large and polygonal in shape with abundant, eosinophilic, and finely granular cytoplasm. The final pathologic examination reported well-differentiated (grade 2) pancreatic endocrine tumor and metastasis in the liver. This was supported by the immunohistochemical study that showed the tumor cells were positive for neuron-specific enolase, chromogranin, and synaptophysin (Fig. 2). The tumor had 5% Ki-67-positive cells, and the number of mitoses was 1/10 high-power fields. The 24-hr urine 5-hydroxyindoleacetic acid levels were 10.4 mg/day (normal, < 10 mg/day). The patient had normal levels of serum chromogranin A (37.6 ng/mL; normal, 27-94 ng/mL), parathyroid hormone (51.2 pg/mL; normal, 13-60 pg/mL), and gastrin (74.5 pg/mL; normal, 0-100 pg/mL).
Interferon alpha (6 million IU) was administered subcutaneously three times a week as first-line therapy. Subsequently, a combination etoposide/cisplatin chemotherapy, a somatostatin analog (Sandostatin® LAR, 1 million IU), and gemcitabine monotherapy were administered; these therapies, however, failed to demonstrate tumor shrinkage or stabilization. Therefore, high-intensity focused ultrasound ablation (HIFU) with palliative intent was performed for nodules in segments 6 and 8. However, 2 months after HIFU treatment, the size of the hepatic nodules increased. The disease status rapidly deteriorated, but the patient's general performance was sufficient to allow considering further chemotherapy. We chose sorafenib (800 mg/day, daily) as the salvage treatment. DNA from formalin-fixed, paraffin-embedded primary tumors was used for BRAF mutation analysis. The mutation in exon 15 of the BRAF gene was not found. After 3 weeks of sorafenib treatment and two episodes of grade 3 skin toxicity, the dose was reduced to 600 mg/day. When the skin toxicity resolved to less than grade 1, we increased the dose of sorafenib to 800 mg/day; the skin toxicity did not increase and did not limit the dose. Our patient tolerated sorafenib well. A follow-up contrast-enhanced CT scan after 2 months of sorafenib therapy revealed that the lobulated mass in the uncinate process had become smaller; the maximal diameter was now 5.4 cm (Fig. 1B). Post-chemotherapy, a contrast-enhanced CT scan showed that the target liver lesions had a density of 16 Hounsfield units. Thirteen months after beginning sorafenib, no disease progression was observed and the patient was well.
DISCUSSION
Pancreatic endocrine tumors account for less than 5% of pancreatic cancers (1). They are believed to be derived from amine precursor uptake and decarboxylation stem cells and arise from cells producing insulin (B cells), glucagon, somatostatin (D cells), and pancreatic polypeptide (5). Pancreatic endocrine tumors are histologically classified on the basis of tumor cell differentiation as well differentiated endocrine tumors (benign or low-grade malignancy), well differentiated endocrine carcinomas, poorly differentiated endocrine carcinomas (small-cell carcinomas), mixed exocrine and endocrine carcinomas (such as adenocarcinoids), and several extremely rare neuroendocrine-like lesions (5). Immunohistochemically, these tumors are positive for markers of neuroendocrine tissue including neuron-specific enolase, chromogranin, and synaptophysin (6). Well differentiated endocrine tumors/carcinomas of the pancreas, as in our patient, stain abundantly and diffusely for both chromogranin and synaptophysin, whereas poorly differentiated endocrine carcinomas stain abundantly and diffusely only for synaptophysin. Pancreatic endocrine tumors are clinically classified as being functioning or non-functioning. The incidence rate of non-functioning pancreatic endocrine tumors is between 15% and 82%; they are pancreatic tumors with endocrine differentiation that lack a clinical syndrome of hormone hypersecretion (7, 8). Clinical presentation of non-functioning pancreatic endocrine tumors is related to the mass effect of the tumor, and symptoms resemble those of pancreatic adenocarcinoma; these include jaundice, abdominal pain, weight loss, and the appearance of an abdominal mass. Radiologically, non-functioning tumors are usually manifested as large well-defined masses with moderate to strong enhancement without invasion of adjacent vessels (9). In our case, the lesion showed a large enhancing pancreatic mass with no invasion of adjacent vessels and the common bile duct; cystic degeneration of multiple hypervascular metastases was observed. These findings were suggestive of non-functioning endocrine carcinoma rather than pancreatic adenocarcinoma.
The median survival time of patients with metastatic pancreatic endocrine tumors was only 23 months, but the recent impact of multimodality treatments has prolonged the survival duration to a median of 70 months (8). Poor prognosis is associated with a high-grade tumor, a high mitotic rate, the presence of necrosis, and lymph node and liver metastasis. No difference in survival duration exists between patients with functioning and non-functioning forms of the tumors (2). Surgical resection is considered to be the optimal treatment for localized disease. It remains the only curative treatment with a reported 5-yr survival rate for 75% of cases of non-functioning tumors (10). In patients with locally advanced and surgically unresectable non-functioning tumors in the absence of extrapancreatic metastatic disease, the appropriate management remains a difficult therapeutic dilemma due to the indolent natural history; median survival is approximately 5 yr. However, more than 70% of patients with pancreatic endocrine tumors have metastatic disease at the initial presentation (7). In these patients, trials of immunotherapy and cytotoxic chemotherapy have established the activity of several drugs, including interferon alpha, a somatostatin analog, streptozocin, doxorubicin, fluorouracil, dacarbazine, temozolomide, and combinations of these modalities. Streptozocin-doxorubicin combination chemotherapy has been considered the standard of care for patients with metastatic, non-functioning tumors (4). However, relatively few patients with pancreatic endocrine tumors participated in the clinical trials due to the low incidence of the disease, the performance status of the patients and their previous therapeutic management, and the heterogeneity and stage of progression of their tumors. Moreover, retrospective studies have raised questions about the value of streptozocin-based chemotherapy because of a low response rate (11). Hence, more effective and less toxic therapies are clearly needed for metastatic pancreatic endocrine tumors.
The molecular pathogenesis of endocrine tumors of the digestive tract is largely unknown and does not involve mutations in classical oncogenes such as Ras, Myc, Fos, and Jun, or tumor suppressors like p53 and the retinoblastoma susceptibility gene. Recent investigations of neuroendocrine tumors have indeed shown attractive potential molecular targets such as epidermal growth factor receptor (EGFR) (12), vascular endothelial growth factor (VEGF) and its receptor (VEGFR) (13), insulin-like growth factor receptor (IGFR) (14), and mammalian target of rapamycin (mTOR) pathway kinases (15). These findings suggest a role for molecular targeted agents in treatment of these tumors, and several targeted agents directed against neuroendocrine tumors are currently in clinical trials. In a phase II trial of imatinib therapy for carcinoid tumors, Carr et al. (16) reported that only one of 27 patients achieved an objective response, but a significant number of patients with progressive disease achieved stabilization and an encouraging median progression-free survival duration of 24 weeks. In a phase II study to evaluate the efficacy and safety of sunitinib, the overall objective response rate in patients with unresectable neuroendocrine tumors was 16.7% (11 of 66 patients), and 68% (45 of 66 patients) had stable disease (17). Everolimus, an inhibitor of mTOR, showed an overall objective response rate of 9.6% in 115 patients with carcinoids or islet cell tumors in a phase II trial (18).
The mitogen-activated protein kinase (MAPK) pathway is pivotal for the regulation of proliferation and protection from apoptosis in many cell types. In endocrine and melanoma cells, MAPK is activated by the Raf kinases B-Raf and Raf-1, which are in turn stimulated by small G-protein Rap1. B-Raf is highly expressed and has been shown to be the main activator of MAPK signaling in neuroendocrine cells (19). These findings imply a possible role for targeted agents against B-Raf for the treatment of pancreatic endocrine tumors. However, no published clinical data exist on the use of a B-Raf inhibitor as therapy for patients with these tumors. The orally administered targeted-agent sorafenib (Nexavar®, Bayer Pharmaceuticals Corporation, West Haven, CT, USA) was approved for patients with advanced renal cell carcinoma and hepatocellular carcinoma. It inhibits tumor-cell proliferation and tumor angiogenesis, and increases the rate of apoptosis in a wide range of tumor models. It acts by inhibiting the RAF serine/threonine kinases (RAF-1, wild-type BRAF, V600E BRAF), the receptor tyrosine kinase activity of VEGFRs 1, 2, and 3, and platelet-derived growth factor receptor β (PDGFR-β). A study by Wilhelm et al. (20) demonstrated that sorafenib inhibited the MAPK pathway in neuroendocrine cell lines; MAPK activation and proliferation were inhibited by high concentrations of sorafenib. Nonspecific effects of sorafenib such as the inhibition of the tyrosine kinase receptors of VEGFR, PDGFR-β, and c-KIT may have the potential to increase the effect of specific Raf kinase inhibition in a clinical setting. The efficacy of sorafenib was also observed in wild-type and mutant BRAF.
The present patient had an advanced pancreatic endocrine tumor and his clinical features suggested a poor outcome. The tumor was progressing and showed no response to biotherapy and conventional cytotoxic chemotherapy. We used sorafenib as a salvage alternative. The overall response to sorafenib was stable. Since beginning targeted therapy and up to the time of this report, the patient has received sorafenib and remains well without progression of disease. Stabilization of disease without tumor shrinkage may represent a meaningful benefit and has become an important issue since the introduction of molecular targeted agents in clinical trials.
In conclusion, this is the first case report indicating that sorafenib may be effective against pancreatic endocrine tumors. Further clinical studies are needed to explore sorafenib efficacy in the treatment of these tumors.

 XML Download
XML Download