

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Idiopathic hypoparathyroidism is an endocrine disorder characterized by hypocalcemia and hyperphosphatemia due to impaired secretion of parathyroid hormone (PTH). Molecular genetic studies have elucidated the dynamic pathogenesis of idiopathic hypoparathyroidism. It may occur as part of a polyglandular autoimmune disorder or as a complex congenital defect, such as in DiGeorge's syndrome. In addition, it may occur as a solitary endocrinopathy which is called isolated hypoparathyroidism. Isolated hypoparathyroidism may be caused by a mutation in the PTH gene, glial cells missing (GCM)2 gene or calcium-sensing receptor (CaSR) gene (1, 2).
Gain-of-function mutations in the CaSR gene cause familial hypocalcemia (3). Here we report a Korean family with two affected siblings and their phenotypically silent father, who were found to carry mutations in the CaSR gene. This case is the first report in Korea.
CASE REPORT
A 24-yr-old woman was seen in the endocrinology clinic at Samsung Medical Center because of transient numbness and periodic paralysis. The patient reported that the symptoms started 10 yr ago. The patient experienced occasional, brief episodes of paralysis during exertion that resolved with rest. Mild numbness and tingling of the hands and feet were also present intermittently.
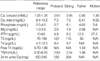
On examination, the patient appeared well. Her vital signs were normal; her height was 155 cm, and her weight was 44 kg. Neurologic examination was significant for positive Trousseau and Chvostek signs. The remainder of the physical examination was normal. Laboratory tests revealed hypocalcemia (7.3 mg/dL; reference range 8.4-10.2), hyperphosphatemia (5.7 mg/dL: reference range 2.5-4.5), decreased 1,25-dihydroxycholecalciferol ([1,25(OH)2D] 13.2 pg/mL: reference range 25.1-66.1), and decreased iPTH (4.9 pg/mL: reference range 10-65). 25-hydroxycholecalciferol ([25(OH)D] 20.2 ng/mL: reference range 11-70) and 24-hr urinary calcium excretion was normal as were bone densitometry, thyroid functions tests, and radiographs of the kidney, ureter, bladder (KUB) and skull. The patient was treated with calcium carbonate and alfacalcidol with resolution of symptoms and dosages were adjusted to maintain a serum calcium level within the lower end of the normal reference range.
The older brother of the proband had a history of generalized seizures since he was 20-yr-old for which he was seen by a neurologist at an outside hospital. He also presented to Samsung Medical Center with his sister because of intractable seizure. Initial evaluation revealed a serum calcium concentration of 7.5 mg/dL (reference range 8.4-10.2), a serum phosphorus concentration of 6.1 mg/dL (reference range 2.5-4.5), and a serum magnesium concentration of 1.9 mg/dL (reference range 1.9-2.5). The serum concentration of iPTH level was 6.2 pg/mL (reference range 10-65). He was treated with an antiepileptic medication and calcium carbonate, but seizure activity persisted. He was taking calcium carbonate 3 g per day with antiepileptic drug. He was admitted to the neurology ward where he underwent EEG and brain imaging. The laboratory test on admission showed a serum calcium concentration of 7.1 mg/dL (reference range 8.4-10.2), a serum phosphorus concentration of 5.6 mg/dL (reference range 2.5-4.5), and a serum ionized calcium concentration of 0.92 mM/L (reference range 1.05-1.35). Brain magnetic resonance imaging showed non-physiologic calcifications in the basal ganglia, bilateral frontal lobes, and cerebellum. The EEG was normal. A dosage of calcium supplement was adjusted, and alfacalcidol was added. He reported subsequent absence of seizure activity during follow-up. During follow-up the calcium level increased up to 8.3 mg/dL (reference range 8.4-10.2) and the ionized calcium level increased up to 1.0 mM/L (reference range 1.05-1.35). After seizure activity subsided, he is followed-up by the physician near the home.
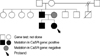
Although the parents of patients denied symptoms attributable to hypocalcemia, they agreed to evaluation. Laboratory examination of their father revealed hypocalcemia, hyperphosphatemia, and an inappropriately low PTH level. The results of laboratory test are shown on Table 1. The mother's laboratory work-up was normal. The remaining members of the family were not included in this study because of inaccessibility (Fig. 1).
DNA analysis
After we obtained informed consent, the proband and her family members were examined to detect the CaSR mutations by direct sequencing analysis. Genomic DNA was extracted from blood using a Wizard genomic DNA purification kit (Promega, Madison, WI, USA). Each of the 6 exons of the CaSR gene were amplified by polymerase chain reaction using appropriate intronic primer sets designed by the authors (Table 2). Cycle sequencing was performed with a BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, USA) on the ABI-3100 Genetic Analyzer (Applied Biosystems).
Direct sequencing showed that all affected family members have a C to T transition at nucleotide 662 resulting in a Pro221Leu missense mutation in exon 3 of the CaSR gene (Fig. 2).
DISCUSSION
In recent years, important advances in endocrinology have resulted from the application of the methods of molecular biology. These advances have helped not only to demonstrate the roles of mutant genes in the etiology of some inherited disorders, but have also helped to identify the chromosomal locations of susceptibility genes that predispose individuals at risk for a particular disorder such as familial hypocalcemia. The results of the studies on the molecular genetics of the hypoparathyroid disorders have helped to elucidate some of the underlying pathways that are involved in calcium homeostasis (1).
Theoretically, hypocalcemic disorders can be caused by deficiency of PTH, by a defect in the PTH/PTHrP receptor, or by insensitivity to PTH caused by defects downstream of the PTH/PTHrP receptor. The mutations in several genes such as CaSR (4), PTH (5), GCM2 and GATA3 (6) have been shown to cause hypoparathyroidism. Among them mutations in CaSR and PTH show autosomal dominant inherited forms of isolated hypoparathyroidism. GATA3 mutation is inherited autosomal dominantly, but it usually accompanied by multiple anomalies such as deafness and renal impairment (1, 2). Proband and her family member had hypocalcemia in conjunction with low PTH level, and a defect in the PTH/PTHrP receptor and insensitivity to PTH could be excluded. The absence of associated anomalies could partly exclude the mutations in GCM2 and GATA3 genes.
In 1993, it was discovered that the mutation of the CaSR gene causes familial hypocalciuric hypercalcemia and severe neonatal hyperparathyroidism. Subsequently, it was learned that a loss-of-function mutation causes hypercalcemia and gain-of-function mutation causes hypocalcemia (4), which is called autosomal dominant hypocalcemia (ADH).
The CaSR gene is located on chromosome 3q13.3-q21, and it encodes a cell surface protein of 1078 amino acids that is expressed in the PTH-producing chief cells of the parathyroid glands and also in cells lining the kidney tubule. The CaSR belongs to the family of G-protein-coupled receptors (3, 7, 8). The CaSR in the parathyroid gland detects small changes of extracellular calcium concentration and regulates transcription of the PTH gene. The CaSR that is expressed in the distal renal tubule regulates renal calcium excretion (1). Thus, the CaSR is vital to maintaining normal serum calcium levels (9). Loss-of-function mutations in the CaSR gene are associated with a severe neonatal hyperparathyroidism and familial hypocalciuric hypercalcemia. Gain-of-function mutations in the CaSR gene are associated with a familial hypocalcemia (3). Since Pollack et al. reported an activating mutation in the N-terminal, extracellular domain of the CaSR gene (4), several activating mutations in the CaSR gene have been reported (9). Currently, there is a database of mutations of CaSR available on the internet (http://www.casrdb.mcgill.ca/) which lists all the known CaSR mutations and their effects (10). Pro221Leu missense mutation was first reported in 2000, and the mutation is located in a lesion of the extracellular domain of the receptor. Replacement of a large, rigid proline with a smaller and more flexible leucine residue, potentially changing the conformation of the calcium-binding region to resemble that of bound calcium or facilitating the binding of calcium ions, resulting in an active mechanism (11). Four more cases were reported in CaSR database after 2000.
It is important to distinguish hypocalcemia due to an activating mutation of CaSR from isolated hypoparathyroidism because ADH patients can develop nephrocalcinosis and renal impairment during treatment with calcium and vitamin D. The distinction between isolated hypoparathyroidism and ADH may be difficult to make on based on clinical information alone. Therefore, mutational analysis of the CaSR gene can be considered to assess the risk of nephrocalcinosis during treatment of PTH-deficient hypoparathyroidism (12).
In conclusion, although a case of familial benign hypocalciuric hypercalcemia due to a loss-of-function mutation in the CaSR gene has been reported in Korea (13), a gain-of-function mutation of the CaSR gene that causes familial hypocalcemia has not yet to be reported in Korea. Here we have described the first Korean family with ADH with a review of the literature.


 XML Download
XML Download