

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Oral antiviral agents are currently available and are the main option for the treatment of patients with chronic hepatitis B. In a large proportion of patients, antiviral therapy can slow down the disease progression by hepatitis B virus (HBV) DNA suppression and hepatitis 'e' antigen (HBeAg) seroconversion. However, the potential emergence of drug resistant mutations remains a caveat of oral antivirals; mutations increase as the treatment duration extends (1, 2). In addition, mutations to one drug may lead to resistance to other drugs. Patients with lamivudine (LMV)-resistant rtM204I/V (±rtL180M) mutations in the catalytic tyrosine-methionine-aspartate-aspartate (YMDD) motif are less responsive to entecavir (ETV) (1, 2). Among adefovir (ADV) treated patients, rtA181T or rtA181V mutations have been associated with a 2-6 fold decrease in sensitivity to LMV (2).
Mutation of the HBV polymerase can alter the amino acid codon of the surface genes and vice versa, because the polymerase gene, the main target of antiviral drug resistant mutations, has overlapping reading frames with the surface region (1, 3). In LMV treated patients, the P120A mutation in the S region results in hepatitis B surface antigen (HBsAg) detection failure (4). Lee et al. (5) reported overlapping surface gene mutations of HBV in an LMV-treated patient who finally lost HBsAg. In this patient, they detected deletions of nucleotides 23 to 55 (amino acids 12 to 22) in the Pre-S2 region, and amino acid substitutions at I126S, T131N, M133T, and S136Y in the 'a' determinant region of HBsAg.
Overlap of surface gene mutations, such as sW172stop in patients carrying rtA181T, and sL173F in patients carrying rtA181V, have been reported in chronic hepatitis B patients receiving ADV treatment. However, the clinical consequences of these changes have not been determined (1, 2). The goal of this study was to evaluate the surface gene sequence in ADV-resistant patients carrying A181T/V mutations and to determine the associated clinical significance.
MATERIALS AND METHODS
Patients
Twenty-two patients with chronic hepatitis B were included in this study. Nine patients had not been previously treated with antiviral agents and thus formed the control group (nucleos[-t]ide naïve patients, Group C). The remaining thirteen patients were ADV-resistant, having an rtA181T mutation, rtA181V mutation, or both mutations (ADV polymerase mutation group, Group P). These patients developed ADV resistance while undergoing ADV monotherapy as rescue therapy for LMV-resistant HBV (rtM204I/V±rtL180M). The ADV-resistant mutations were detected using restriction fragment mass polymorphism (RFMP) method as previously described (6). Serum was collected from the patients every two to three months during treatment and was kept at -20℃ until the mutation analyses were performed. The serum collected at the time of ADV-resistant rtA181T/V mutation detection was used for sequencing analysis. Informed consent was obtained from each patient and the experimental protocol was approved by the Korea University Guro Hospital Human Research Committee.
Biochemical and virology monitoring
At the time of serum collection, we conducted routine full blood examinations and liver function tests, including tests for aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, albumin, prothrombin time, blood urea nitrogen (BUN), and creatinine. HBsAg and antibody to the HBs antigen were measured using the commercially available radioimmunoassay kits (Abbot Laboratories, North Chicago, IL, USA). The HBsAg titer was measured using the chemiluminescent microparticle immunoassay (CMIA) ARCHITECT® HBsAg assay (Abbot Laboratories). HBeAg and antibody to the HBe antigen were measured using commercially available radioimmunoassay kits (Shin Jin Medics Inc., Goyang, Korea). HBV-DNA was quantified using the Hybrid Capture II assay (Digene Diagnostics Inc., Beltsville, MD, USA) or the VERSANT® HBV DNA 3.0 assay (Siemens Healthcare Diagnostics Inc., Tarrytown, NY, USA) and data showed 'pg/mL' were converted to 'copies/mL' (1 pg=283,000 copies).
Polymerase chain reaction
Hepatitis B viral DNA was extracted from 200 µL of serum using the QIAamp® MinElute® Virus Spin kit (Qiagen Inc., Vanencia, CA, USA) according to the manufacturer's instructions. The S coding region of HBV was amplified using HBS-1S (the forward primer) and HBS-1AS (the backward primer) as previously described (5). The amplification conditions consisted of 94℃ for 3 min, 35 cycles of 94℃ for 30 sec, and 55℃ for 30 sec. This was followed by a final primer extension at 72℃ for 1 min using the Taq PCR Core Kit (Qiagen Inc.). The polymerase chain reaction (PCR) amplified HBV DNA was purified using the QIAquick® PCR Purification Kit (Qiagen Inc.) according to the manufacturer's instructions.
Sequencing
The primers used for direct sequencing were HBS-1S, HBS-1AS, HBS-2S, and HBS-3S as previously described (5). Sequencing reactions were performed in an MJ Research PTC-225 Peltier Thermal Cycler using an ABI PRISM BigDye™ Terminator Cycle Sequencing Kit with AmpliTaq DNA polymerase (Applied Biosystems Inc., Foster, CA, USA) according to the manufacturer's instructions. Single-pass sequencing was performed on each template using the aforementioned primers. The fluorescent-labeled fragments were purified from the unincorporated terminators with an ethanol precipitation protocol. The samples were resuspended in distilled water and subjected to electrophoresis in an ABI PRISM 3730XL Analyzer (Applied Biosystems Inc.).
Analysis
The nucleotide sequences were compared with those of the HBV genotype C subtype 'adr' registered at the National Center for Biotechnology Information (NCBI, nucleotide LOCUS AF286594). The genotype of the obtained HBV DNA sequences was determined by a web-based genotyping tool for viral sequences at the NCBI (7).
Statistical analysis
We conducted the Fisher's exact tests for categorical variables and Mann-Whitney's U-tests for continuous variables to compare patients with the rtA181T/V mutation to those without the rtA181T/V mutation. Values are expressed as median (range). A P value <0.05 was considered statistically significant.
RESULTS
Demographic and biochemical characteristics
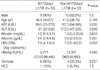
Demographic and biochemical data are summarized in Table 1. Among the 13 patients in Group P, the majority were males (n=10). All patients in Group C were males. Biochemical data are represented as median (range) (median of Group P vs. Group C, P value). Group P was significantly older than Group C, 45 (28-75) (47 vs. 39, P=0.025), had higher levels of albumin 4.2 (2.5-5) (4.2 vs. 3.6, P>0.05). Group P also had lower levels of AST 100.5 (23-385) (65 vs. 193, P=0.001), ALT 155 (22-385) (78 vs. 262, P<0.001), and total bilirubin 1.1 (0.4-33.6) (1.0 vs. 1.3, P>0.05). Ten of the 22 patients had liver cirrhosis (LC) (Group P=7, Group C=3). Thirteen of the 22 patients were HBeAg positive (Group P=7, Group C=6). Two cases of hepatocellular carcinoma (HCC) were included (Group P=1, Group C=1).
HBV genotype, subtype, and point mutations
All patients in our study were infected with HBV genotype C. Twenty-one of the 22 patients had the 'adr' subtype and one patient had the 'ayr' subtype. For the Pre-S1, Pre-S2, and S genes, the incidence of mutations in Group P and Group C were 3 vs. 3 (P>0.05), 1 vs. 3 (P>0.05), and 4 vs. 2 (P>0.05), respectively. The median number of point mutations in the entire region was 8 (Group P=8, Group C=10, P>0.05). Most cases had mutations in the S gene except for one case in Group C. One patient in Group P did not have a mutation in the Pre-S1 gene and seven patients (Group P=5, Group C=2) had no mutations in the Pre-S2 gene (Table 2).
Changes in the serum hepatitis B surface antigen titer
We conducted serum HBsAg titers to evaluate the clinical consequences of the surface gene mutation. The median HBsAg titer was 7,590 IU/mL (Group P=5,950, Group C=15,090, P>0.05). There was no significant difference, regardless of whether they harbored the rtA181T/V mutation (Table 2).
Pre-S1 region
In the Pre-S1 region, 12 patients (six cases in each group) had nucleotide deletions at position '21' (amino acid [a.a.] 7) and 18 patients (nine cases in each group) had nucleotide deletions at position '30' (a.a. 10). Nucleotide deletions at position '59' (a.a. 20) were detected in 5 of 13 patients in Group P and 2 of 9 patients in Group C. An H51Q mutation was detected in two patients (one case in each group). Eight of 13 patients in Group P and 3 of 9 patients in Group C had a V60A mutation. G2A (n=1), G73D (n=1), L74I (n=2), A91T (n=1), and A95T (n=1) mutations were detected in Group P. In Group C, A81T (n=1), T86A (n=2), and A95V (n=1) mutations were detected. Three cases of I84T mutations were found in Group C, compared to only one in Group P (Fig. 1).
Pre-S2 region
A start codon mutation was identified in five patients (Group P=3, Group C=2) in the Pre-S2 gene. All five patients with this mutation had cirrhosis, and two of these patients also had HCC (Group P=1, Group C=1). V32A, I45T, and T49I mutations were detected in the patients with HCC, both groups equally. An F46S mutation was observed in Group P (n=2); a Q2K mutation in Groups P (n=1) and C (n=1); an A11T mutation in Groups P (n=2) and C (n=1); and an F22L mutation in Groups P (n=2) and C (n=2).
In Group C, there was one case of each of the following mutations: N4K, T7A, H9Q, R16P, R16G, V17P, V17A, R18S, G19S, L20Q, P54Q, nucleotide 418-423 deletion (a.a. 21-22), 419 deletion (a.a. 21), and 425 deletion (a.a. 23). In Group P, mutations I42T, P54L, and nucleotide 406 deletion (a.a. 17) were detected (Fig. 2).
S region
Among the patients in Group P, sW172stop (n=5) and sL173F (n=5) mutations were detected. In the S gene, an A184V mutation was found in 9 of 13 patients in Group P. However, it was not detected in Group C (P=0.002). Other mutations that we detected in the S gene include S3N (Group P=4, Group C=2), I68T (Group P=3, Group C=1), I126T (Group P=4, Group C=1), and L213I (Group P=6, Group C=1). These mutations were most frequent in Group P.
In Group C, there was one case of each of the following mutations: G10stop, L15V, I28V, W36L, F41S, T47A, Q51L, N59H, C69stop, L87V, F93L, V96G, L97P, G102C, T131P, Y200F, and C221Y. Similarly, in Group P, there was one case of each of the following mutations: F41C, A45V, P49L, Q54L, P62L, I92T, L98V, T140S, A157V, W182stop, and S204R. An L21S mutation was detected in Groups P (n=2) and C (n=2), and R24Q and P203R mutations were found in Groups P (n=1) and C (n=1) (Figs. 3, 4).
Entire amino acid changes in both groups
Among these mutations, only the W172stop/L173F (P<0.001) and A184V (P=0.002) mutation were significantly different between the groups (data not shown).
Comparison of demographic, biochemical and clinical characteristics between patients with and without the W172stop/L173F mutation
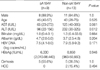
We also compared patients both with and without the W172stop/L173F mutation (Table 3). AST (P=0.035) and ALT (P=0.009) levels were significantly lower in patients with W172stop/L173F mutation. However, there was no significant difference when the same comparison was done in Group P alone.
Comparison of demographic, biochemical and clinical characteristics between patients with and without the sA184V mutation
We compared patients both with and without the sA184V mutation (Table 4). Only the ALT level was significantly lower in patients with the sA184V mutation (P=0.012). However, there was no significant difference when the same comparison was done in Group P alone.
DISCUSSION
HBV is an enveloped virus and contains a partially double-stranded DNA genome of approximately 3,200 base pairs (1, 3, 8). The major envelope protein of HBsAg consists of 226 amino acids and is coded by the S gene (9). The polymerase gene overlaps all six other genes, including the three envelope genes, Pre-S1, Pre-S2, and S, which encode for the large (by the Pre-S1/S2/S gene), middle (by the Pre-S2/S gene), and small (the S gene) envelope proteins, respectively (1, 10). Due to the overlap between the polymerase and surface genes, the selection of a drug-resistant HBV polymerase gene mutant may have important clinical and diagnostic implications (1, 3).
Deletions and mutations of overlapping surface genes have been reported in LMV treated patients who loose HBsAg (3). In addition to surface gene deletions and mutations, deletions of nucleotides in the Pre-S2 region and amino acid substitutions in the 'a' determinant region of HBsAg have been reported (5). The LMV-resistant polymerase mutation, rtV173L (sE164D), has been reported to reduce the antigenicity of HBsAg (11).
Drug resistant mutations to ADV have been reported mainly in the HBV polymerase domain D rtN236T or the domain B rtA181V/T (1, 12). Whereas a domain D rtN236T mutation does not overlap with the envelope gene, a mutation at rtA181T can result in a stop mutation in the envelope region of the S gene (sW172stop). In addition, an ADV-resistant mutation at rtA181V results in a concomitant change at sL-173F (1). Therefore, we selected LMV-resistant chronic hepatitis B patients who had developed ADV-resistant A181T/V mutations during ADV rescue therapy. We sought to detect other mutations caused by the ADV-resistant mutation and its clinical implications. Patients who had not received antiviral treatment were selected for the control group.
In the present study, biochemical data showed that AST, ALT and total bilirubin levels were higher in Group C. And it can be considered that Group C has relatively worse biochemical level. One limitation should be noted: we used indirect markers or surrogates of liver function in the biochemical tests and fibrosis, proliferation, apoptosis, and other pathogenic processes were not measured. The patients should be followed for a long period of time and the clinical impact of the rtA181T/V mutation should be evaluated.
The results of our study showed that treatment induced or naturally occurring mutations of the Pre-S1, Pre-S2, and surface antigen regions were detected in most of the patients studied. In Group P, although statistically insignificant, Pre-S1 region nucleotide deletions at position '59', and mutations V60A and L74I were the primary changes detected. Five patients carrying Pre-S2 start codon mutations had either cirrhosis or HCC. In addition, V32A, I45T, and T49I mutations at Pre-S2 were detected in the HCC patients only. An F46S mutation in the Pre-S2 region was detected only in patients with the ADV-resistant rtA181T/V mutation. These results are consistent with a number of previous reports indicating that Pre-S mutations are associated with advanced liver disease and active viral replication (13-19). In Group C, a number of point mutations were detected. A similar pattern of mutations was also detected in Group P. There were no specific mutations confined to the Pre-S2 region in Group C. Nucleotide 21 deletions were frequently observed in both Groups P and C, and nucleotide 30 deletions were observed in all but four cases. We are uncertain as to what caused the sequence changes. It is difficult to suggest that the sequence changes were caused by resistant mutations because these deletions were more common in Group C. The development of Pre-S deletion mutants depends on HBV genotypes (20). Recently, one report showed that Pre-S deletions were more common in Korean HBV genotype strains and correlated with genotype C and advanced liver disease (21). The mutations in the Pre-S region, particularly the deletions, may affect the ratio between the small and large envelope proteins, resulting in endoplasmic reticulum stress associated with the aggravation of liver disease (21, 22). These patients should be observed closely.
The S gene mutation, A184V, was detected in the ADV-resistant rtA181T/V polymerase mutation group alone. This mutation site overlaps with the rt192 region in the polymerase gene. However, the A184V mutation did not result in amino acid changes in the polymerase region. There is one published report that the A184V mutation was related to reduced or negative HBsAg signal (23). In our study, however, changes in HBsAg titer were not observed. When compared with patients who have not A184V mutation including Group C, ALT level alone was significantly lower in patients with A184V mutation. But it is difficult to conclude that it is caused by A184V mutation, because this difference was no longer significant when the analysis was done in pure Group P and patient's number was not enough. The clinical implications of this remain to be determined. Of all mutations within the 'a' determinant region of HBsAg, the most common antigenic determinant mutations are in amino acids 124 to 147 (24). Substitutions within the 'a' loop can create a putative glycosylation site in mutant HBVs that result in undetectable HBsAg antigenicity (25). In our study, an I126T mutation was detected in 4 of 13 ADV mutants and one of nine control cases. One case of a T131P mutation in Group C and one case of a T140S mutation in Group P were also detected. Some investigators have reported that s126 threonine is a wild type variant and nucleotide substitution of threonine has no specific effect (26-29). To determine the clinical significance of I126T, T131P, and T140S mutations, further studies are required. It is possible that these amino acid substitutions were caused by a remaining LMV-resistant mutation, though a rtM204I/V mutation was not detected in sequencing analysis.
Although surface gene mutations were common in Group P, serum HBsAg titers were not significantly different from those of Group C. Despite the fact that we did not measure in vitro hepatitis B surface antigen production, the impact of coexisting wild type virus, viral replication, fitness, packaging, and antigenic targets for immunoassay should be considered. Full-length HBV genomes should be cloned and the S gene should be sequenced and analyzed. It should be noted that, in this study, patients who were nucleoside treatment-naïve controls had a variety of Pre-S gene mutations as well as surface gene mutations.
We detected sW172stop and sL173F mutations in 10 of the 13 patients with ADV-resistant rtA181T/V polymerase mutations. A large percentage of the cases with rtA181T mutations developed sW172stop mutations. In cases with ADV treated LMV-resistant mutations, the rtA181T mutation was reported at the ADV treatment baseline with low HBV DNA titers (30).
In conclusion, the results of this study showed that treatment-induced or naturally occurring mutations of the Pre-S1, Pre-S2 and surface antigen regions were frequent. The sA184V mutation was detected in the A181T/V polymerase mutation group alone. Although sW172stop and sL173F mutations were detected in the polymerase mutation group, a reduction in HBsAg titer was not found. Further study is needed to determine the clinical implications of viral replication, topographic alteration, fitness, envelope gene production, and interaction with wild type HBV DNA.






 XML Download
XML Download