

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The congenital long QT syndrome (LQTS) is a clinical disorder of genetic origin. It is characterized by abnormal QT interval prolongation (rate-corrected QT or QTc) and susceptibility to sudden death owing to a specific type of polymorphic ventricular tachycardia, torsade de pointes (1).
Two forms of inherited LQTS have been described: Romano-Ward syndrome (RWS), an autosomal dominant inherited disorder with prolongation of the QTc without other associated noncardiac abnormalities (1), and Jervell and Lange-Nielsen syndrome (JLNS), an autosomal recessive disorder with LQTS associated with sensorineural deafness (2).
JLNS usually results from homozygous loss-of-function mutations in either KCNQ1 or KCNE1. Patients with JLNS have a more severe cardiac phenotype than those with RWS. Furthermore, JLNS accompanies complete loss of Iks in hair cells and endolymph of the inner ear, which result in congenital deafness (3). In this report, we describe the first case of genetically confirmed JLNS in Korea with compound heterozygous mutations in KCNQ1. In addition, one of the mutations is a novel splicing one, which has never been reported before.
CASE REPORT
A five-year-old boy visited the hospital due to recurrent syncope on April 17, 2006. The first episode had occurred at the age of three. It involved seizure-like movement such as tonic-clonic activity, eyeball deviation, and loss of consciousness. Such episodes happened four times during 16 months. They mostly occurred during walking or exercising, and continued for several seconds to several minutes; but cardiac resuscitation had never been required.
His past medical history revealed that he had undergone cochlear transplantation due to congenital sensorineural deafness.
The patient showed no abnormality during physical and neurologic examination. There was no abnormality observed in the electroencephalogram. His resting 12-lead electrocardiogram (ECG) revealed a markedly prolonged QTc interval of 590 ms (Fig. 1). His ECG showed a structurally normal heart. A 24-hambulatory ECG showed no ventricular arrhythmia. An exercise treadmill test showed a marked QTc prolongation as the test progressed. It also revealed T wave alternans (Fig. 2). At that time, he was not on drugs that could extend the duration of QT. Nor did he have any electrolyte imbalance such as hypomagnesemia or hypocalcemia. Because the diagnosis of LQTS was established, an intracardiac electrophysiologic study was not performed.
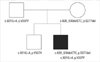
Clinical manifestations of his family members were unremarkable. No family members had experienced syncope, palpitation, epilepsy, or any episode of cardiac arrest. The patient's father and brother had normal QTc intervals, 434 ms and 404 ms, respectively, but his mother had a prolonged QTc interval of 473 ms.
Molecular genetic analysis revealed that the patient had compound heterozygous mutations in the KCNQ1 gene. One was a previously reported deletional mutation, c.828-830delCTC, p.S277del. The other was a novel splicing mutation, c.921G>A, p.V307V (Fig. 3). The allele frequency of the novel sequence variation c.921G>A was 0% in a group of 95 healthy controls. The nucleotide c.921G is located at the end of exon 6; therefore, substitution of G to A was predicted to affect the splicing of mRNA. To determine the effect of this novel substitution, we performed reverse transcription-polymerase chain reaction and detected an abnormally spliced 593-bp mRNA product in which the exon 6 had been skipped (Fig. 3). The subsequent segregation study detected c.828-830delCTC heterozygous mutation in his mother, and c.921G>A in both his father and elder brother (Fig. 4).
At the initial visit, he was clinically diagnosed with JNLS. β-blocker therapy was initiated with oral propranolol (2 mg/kg/d). Since then, he has remained symptom-free for three and a half years, although he has a persistent QT prolongation (QTc=551 ms) as of the most recent ECG.
DISCUSSION
The cardiac delayed rectifier current (Ik) is one of the major determinants of phase 3 repolarization of the cardiac action potential. It is composed of three independent components: an ultrarapid component, called IKur; a rapid component, IKr; and a slow and catecholamine-sensitive component, IKs (4). KCNQ1 encodes the α-subunit and KCNE1 encodes the β-subunit of the channel responsible for the voltage-dependent slowly activating delayed rectifier potassium channel.
Homozygous or compound heterozygous mutation in either KCNQ1 or KCNE1 (minK) causes the Jervell and Lange-Nielsen autosomal recessive form of the disease (JLN1 and JLN2, respectively), which is characterized by cardiac phenotype (long QT interval and susceptibility to ventricular arrhythmia) and sensorineural deafness (5, 6).
JLNS is a very rare disease, found in less than 1% of all patients with LQTS. Moreover, only a few cases with compound heterozygous mutations have been reported (2, 7-9).
In the present study, we described one Korean family in which compound heterozygous mutations in KCNQ1 resulted in JLNS in the proband. Our findings are the first in Korea to demonstrate that compound heterozygote mutations in KCNQ1 causes JNLS. One of two mutant alleles has been associated with LQTS (10) while the other was a novel sequence variant resulting in an abnormal splicing effect of exon 6.
JLNS is the most severe among the major variants of LQTS. Most patients with JLNS become symptomatic and they have a high risk of life-threatening arrhythmias despite treatment with β-blockers (11, 12). Furthermore, JLNS patients suffer from cardiac events in the early period of their lives (11).
The high-risk group of patients with JLNS includes those with a QTc interval exceeding 550 ms or syncope during early childhood (birth to fourth year of life), males less than 20 yr old, and those with KCNQ1 mutation (12). The high-risk patients are strongly recommended implanted cardiac defibrillator (ICD) therapy after the age of five, combined with early medical treatment (12). Although the family members of our JNLS patient who have the same disease do not show any symptoms so far, they may need specific management including ICD therapy or medical treatment to prevent a serious, life-threatening cardiac event. Hence, it is important to perform genetic analysis and to determine whether they have JNLS mutation.
In general, heterozygous carriers of the JLNS mutation are observed to be asymptomatic or mildly symptomatic. One study suggested an explanation for sush an observation that frameshift/truncating mutations are responsible for the majority of JLNS (13, 14). Frameshift/truncating mutations are unable to cause dominant-negative suppression under normal circumstances because they cannot co-assemble with wild-type normal subunits. Therefore, JLNS mutations are milder, and therefore, in the heterozygous form, do little harm. In contrast, the missense mutation in the KCNQ1 gene, which is most common in RWS, can co-assemble with normal subunits, and thereby exert a dominant-negative suppression effect.
Despite limited data, our case also supports the above explanation. The patient's father and brother with a truncating-like mutation have normal QTc interval, while his mother with a missense mutation has prolonged QTc interval.
Therefore, JLNS requires complete gene screening of all family members even if they have normal QTc intervals. Furthermore, the carriers need an individual risk evaluation and careful management depending on the type of mutation. A carrier with a missense mutation should be closely observed.
In conclusion, JLNS has a worse prognosis than any other LQTS genetic groups. The importance of genetic diagnosis has recently been highlighted. Our report identified compound heterozygote deletion/splicing error mutations (c.828-830 delCTC, p.S277del/c.921G>A, p.V307V). To the best of our knowledge, this mutation is the first identified as JLNS in the Korean population. We also suggest early genetic diagnosis for proper management and genetic counseling. Functional assays of the novel mutation were not performed in our study and should be a subject for future research.



 XML Download
XML Download