

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The classification of Ehlers-Danlos syndrome (EDS) is based on the extent of clinical and pathological features. Vascular EDS (type IV EDS, MIM #130050) is the most malignant form and owes its bad reputation to a susceptibility to sudden death from spontaneous catastrophic bleeding or the rupture of blood vessels or hollow organs (1). Type IV EDS is characterized by its clinical manifestations: easy bruising, thin skin with visible veins, acrogeria, and the rupture of arteries, uterus, or intestines (2). Vasculopathy involves the rupture, dissection, or aneurysms of the aorta or large arteries, carotid-cavernous fistula (CCF), and intracranial aneurysms. Arterial complications are the leading cause of death in vascular EDS because they are unpredictable and surgical repair is difficult because of tissue fragility (3). In a recent review of 419 patients, 70% of deaths were the result of arterial rupture, mostly of thoracic and abdominal vessels, but also of cerebral vessels (1). However, bruisability is very striking, and 'ecchymotic-type EDS' is a synonym for type IV EDS. Type IV EDS is inherited in an autosomal dominant fashion but approximately 50% of patients represent new mutations. Most patients have a defect in either the synthesis or structure of type III procollagen, with a genetic mutation in COL3A1. However, it has been suggested that EDS type IV is a genetically heterogeneous disease. Pepin et al. have reported that only 61% of patients with abnormal type III procollagen molecules have mutations in the COL3A1 gene, whereas the others have no mutation in COL3A1 (1). In a recent study, mutations in the genes encoding transforming growth factor β receptors 1 and 2 (TGFBR1 and TGFBR2, respectively) have been identified in patients with a provisional diagnosis of EDS type IV without the characteristic type III collagen abnormalities. This new syndrome was designated Loeys-Dietz syndrome, type 2 (4, 5).
Here, we report our experience of three Korean patients, in whom a clinical diagnosis of EDS type IV was made based on clinical findings. However, a genetic study identified COL3A1 mutations in two of these three patients. In the other one patient, no mutations were observed in COL3A1, TGFBR1, or TGFBR2.
MATERIALS AND METHODS
The diagnoses of these patients were made based on clinical presentation, physical examinations, and radiological findings. Information about deceased family members and other family members who were suspected of being affected was obtained from the probands. Informed consent was obtained.
Genomic DNA was extracted from peripheral blood leukocytes with the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, U.S.A.), following the manufacturer's instructions. The coding exons and their flanking introns of the COL3A1 (total 51 exons) on chromosome 2q31, and TGFBR1 and TGFBR2 on chromosome 9q33-q34 and 3p22, respectively, were amplified using primers designed by the authors (available upon request). Direct sequencing was performed with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, U.S.A.) on the ABI 3100 Genetic Analyzer (Applied Biosystems).
RESULTS
The clinical and pathological findings for the three patients are shown in Table 1.
Patient 1
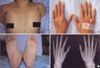
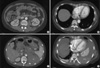
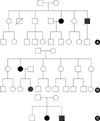
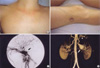
A 48-yr-old woman was admitted with sudden severe epigastric pain radiating to the back one day before admission. Her medical history revealed two uncomplicated cesarean sections. A spontaneous retroperitoneal hematoma had been treated with percutaneous catheter drainage three years previously. On first presentation, her blood pressure was 121/78 mmHg, her pulse rate was 80 beats per minute, and her heartbeat was regular. A clinical examination showed tenderness in the left lower abdominal quadrant, very thin skin with visible vessels on the thorax, which bruised easily with minimal trauma (Fig. 1A), and acrogeria (Fig. 1B, C). Equinus deformity was noted in both feet (Fig. 1C). Acroosteolysis was present (Fig. 1D). The results of a neurological examination were normal. Computed tomography (CT) demonstrated an abdominal aortic dissection with extension from the proximal superior mesenteric artery to both common iliac arteries (Fig. 2A, B). A provisional diagnosis of EDS IV was made. Because of the high surgical mortality rate in patients with EDS type VI, conservative medical treatment was given. Three days later, she suddenly developed back pain with shock. Follow-up CT angiography revealed a progression of the aortic dissection with hemothorax, hemoperitoneum, and retroperitoneal hemorrhage (Fig. 2C, D). The patient died from uncontrolled bleeding before surgical intervention. A family history revealed that her younger brother had died suddenly from cerebral hemorrhage (Fig. 3A).
Genetic analysis revealed a G to T transition (c.2195G>T) leading to the substitution of the glycine at position 732 (p.Gly732Val) of the triple helix of type III protocollagen with valine (Fig. 4).
Patient 2
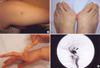
A 36-yr-old woman presented with left ocular pain, headache, diplopia, and tinnitis, which had started one month before presentation. On physical examination, her skin showed a bruise at a trauma site (Fig. 5A). The patient's foot showed chronic dislocation of the first metatarsal joint (Fig. 5B). Hypermobility of the distal interphalangeal joints of the hands was observed (Fig. 5C). Kyphosis of the lumbar and thoracic vertebrae was noted. A family history revealed that her daughter had a chronic subluxation of the proximal interphalangial joint of the left hand (Fig. 3B). Magnetic resonance imaging revealed a large CCF, and the diagnosis was confirmed by angiography (Fig. 5D). The CCF was successfully treated with detachable balloons using a femoral arterial approach. During the procedure, a pseudoaneurysm of the left proximal internal carotid artery occurred, and a stent was inserted. A large subcutaneous hematoma occurred at the puncture site after angiography. Then, surgical evacuation of a massive hematoma was performed, followed by the insertion of a stent graft into the punctured femoral artery, with arteriotomy to control bleeding. The postoperative course was uneventful, except for ophthalmoplegia and the loss of visual acuity due to a neurotropic corneal ulcer. The patient is alive and doing well at the seven-year follow up. A mutation analysis by DNA sequencing of the COL3A1 gene revealed a duplication of 15 base pairs (c.3221_3235dup), which resulted in an interposition of five amino acids (p.Gly1074_Pro1078dup) and a subsequent formation of a repeat unit composed of only two amino acids (Gly-X) (Fig. 6).
Patient 3
Some of the clinical findings of this patient have been reported previously (6). A 21-yr-old woman presented with left ocular pain, severe pulsatile headache, and vomiting after her left eye was hit with a champagne cork. She had bruised easily since she was young. On physical examination, her skin was so thin that the subcutaneous blood vessels were visible (Fig. 7A, B). Echymoses and bruises were noted at trauma sites (Fig. 7B). There was no significant hyperelasticity of the skin or hypermobility of the joints of the hand. Her family history revealed that her younger brother had bruised easily (Fig. 3C). Carotid angiography showed a large CCF and an aneurysm in the cervical portion of the left internal carotid artery (Fig. 7C). Computed tomography of the thoracoabdominal aorta demonstrated a large ovoid aneurysm of the left renal artery and a medium-sized aneurysm of the splenic artery (Fig. 7D). The CCF was successfully occluded with detachable balloons using a femoral arterial approach. Huge groin hematomas and echymoses occurred at the puncture sites after angiography.
Seven years later, she developed a spontaneous hematoma of the left calf, which was relieved by conservative management. The patient is alive and doing well at the eight-year follow up. Genetic analysis revealed no mutation of either COL3A1, TGFBR1, or TGFBR2.
DISCUSSION
Here, we report three patients with a provisional diagnosis of EDS type IV. The first and second patients had mutations of the gene for type III procollagen (COL3A1). Although the other patient showed clinical features compatible with EDS type IV, genetic analysis of COL3A1 did not reveal a mutation. Also, additional genetic analysis of TGFBR1 and TGFBR2 did not show a mutation. Absence of mutations in patients 3 might be either due to a deletion of single or multiple exons in the COL3A1 gene or due to a genetic heterogeneity. Description of patients with EDS type IV-like features in Korea is quite rare (7). To the best of our knowledge, patient 1 and 2 are the first Korean patients with a molecular diagnosis of type IV EDS.
The mutation found in patient 1 is a novel mutation rather than a genetic variation because most of known missense mutations of COL3A1 involves glycine residue and the same variation of the patient was not found in either other family members with normal phenotype or fifty control subjects. Also, any genetic alteration at the duplicated location of patient 2 had not been previously reported in EDS type IV. The nature of the mutation found in patient 2 was quite different from the vast majorities of other previously reported mutations on the COL3A1 gene, which usually were point mutations affecting the glycine residue of the normal Gly-X-Y repeats and caused classical EDS type IV (1). As a result of duplication of 15 base pairs, an interposition of five amino acids among the triple repeats has occurred, and a subsequent formation of a repeat unit consisting of two amino acids (Gly-X) just before the duplicated segment was observed. These atypical properties of the mutation in this patient might have had an effect on her phenotypic features that lacked one of the cardinal manifestations of classical EDS type IV, i.e. thin transparent skin. We previously reported patient 3 as having EDS type IV (6), demonstrating that a genetic diagnosis is mandatory for a final diagnosis in patients with the clinical features of EDS type IV.
Recent developments in genetic diagnosis have broadened the clinical spectrum of type IV EDS. In the largest series of patients with EDS IV, Pepin et al. analyzed the COL3A1 gene in 220 index patients who had biochemical defects in type III collagen (1). They identified causative mutations in the COL3A1 gene in only 135 index cases (61%). In other words, 39% of patients with EDS IV, with abnormal type III collagen, had a mutation in a gene other than COL3A1, which indicates that even EDS IV with a proven defect in type III collagen is a genetically heterogeneous disease. Furthermore, in a recent study, mutations in the TGFBR1 and TGFBR2 genes have been reported in Loeys-Dietz syndrome type 1, which is characterized by the triad of arterial tortuosity and aneurysms, hypertelorism, and bifid uvula or cleft palate (5, 6, 8). Mutations in the gene encoding TGFBR1 or TGFBR2 were also found in 12 (30%) of 40 patients with EDS type IV without the characteristic type III collagen abnormalities. All 12 patients had at least two of the findings associated with EDS IV. They had previously received a provisional diagnosis of EDS IV by a medical geneticist. This new disease was designated Loeys-Diets syndrome type 2, distinguishing it from type 1, which has additional craniofacial abnormalities. Based on these findings, we propose new categories of patients who have EDS-type-IV-like phenotypes (Table 2). Patient 1 and 2 belongs to category I. Patients 3 could be classified into category II or IV. However, there is a possibility that a causative genetic abnormality in patients 3 could be a deletion of single or multiple exons in the COL3A1, TGFBR1 or TGFBR2 gene.
The first patient had the most severe phenotype of the three patients and died of rapidly progressive aortic dissection with rupture. Some authors have suggested that the nature of a mutation relates to the patient's phenotype (9). Mutations in the middle or at the 5' end of a gene produce more variable phenotypes and milder variants compared with those caused by mutations around the 3' end, which have severe manifestations. However, Pepin et al. (1) demonstrated no apparent relationship between the type of complication and the locations of mutations. Even though patient 1 had a substitution in the middle part of the COL3A1 gene product (732nd codon of 1,466 amino acids) and patient 2 had a duplication in the 3' part of the COL3A1 gene (1,074th to 1,078th residues of 1,466 amino acids), patient 1 had relatively severe and full-blown clinical manifestations compared to those of patient 2. The patient 1 survived two cesarean sections. Pepin et al. reported that, among 183 pregnancies with 167 deliveries of live-born infants at term, 12 women died (1). The overall risk of death during delivery was 7%. However, it is not known whether the use of elective cesarean section reduces mortality. The age at death of our patient was 48, which is consistent with a previous report in which the calculated median survival of the entire cohort of 220 index patients was 48 yr (1).
Patients 2 and 3 presented with CCF. CCF is exceedingly rare in the general population, among whom the vast majority is posttraumatic. Spontaneous CCF is one of the most common central nervous system vascular complications of EDS IV. North et al. reviewed these cerebral vascular complications in 202 individuals with biochemically confirmed type IV EDS (10). Of these, six individuals (3%) experienced spontaneous CCF.
One of the striking features of patients 2 and 3 was their vascular complications after invasive vascular procedures. The arteries of both patients were so fragile that they were easily dissected during catheter and guidewire interventions or the ballooning of arteries. They also showed severe complications at transfemoral puncture sites, including giant groin hematoma or severe ecchymosis, which required surgical evacuation of the hematoma and insertion of a stent graft into the injured artery. Therefore, as far as possible, conventional arteriography should be avoided because of the high risk of massive hematoma and/or arterial dissection, as noted in our patients. In an attempt to avoid arterial dissection or rupture, CCF could be closed via a venous rather than the more standard arterial route (11).
In conclusion, we identified three patients with EDS-IV-like phenotypes. Two patients were shown to have mutations in COL3A1. Based on our experience, we emphasize that patients with a presentation suggestive of EDS IV should undergo biochemical analysis of their type III collagen and DNA analysis of the COL3A1, TGFBR1, and TGFBR2 genes. However, these laboratory tests cannot identify the genes causing the category II or IV disease, which await further progress and understanding of the molecular pathogenesis of patients with EDS-IV-like phenotypes.




 XML Download
XML Download