

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Nitric oxide (NO) is one of important mediators of urinary sodium excretion. It may increase the excretion of sodium in the absence of changes in renal hemodynamics (1), suggesting a tonic inhibitory effect on the tubular reabsorption of sodium in kidney. Furthermore, its modulating action on tubular sodium reabsorption is exerted through regulating the membrane sodium transporters.
In the kidney, active reabsorption of sodium occurs in most parts of the tubule through the activity of Na, K-ATPase. It provides the driving force for sodium movement in the basolateral membrane, transporting sodium out of the cell into the interstitium (2). On the contrary, the luminal entry of sodium into the cells occurs through different transporters. Type 3 Na/H exchanger (NHE3) is apically expressed in the proximal tubule (3), whereas the thiazide-sensitive Na/Cl cotransporter (TSC) is involved in apical sodium reabsorption in the distal convoluted tubule (4). In the thick ascending loop of Henle (TALH), Na/K/2Cl cotransporters (BSC1) and NHE3 are mainly responsible for sodium reabsorption (5).
NO has been shown to inhibit both Na/H exchange (6) and Na,K-ATPase activity in proximal tubule cells (7). It has been also demonstrated to inhibit Na-K-ATPase in TALH (8). In the isolated perfused rat TALH, NO donors selectively inhibit the apical BSC1 (8, 9). An altered regulation of these sodium transporters may underlie the salt-sensitive hypertension associated with chronic NO blockade (10-12).
The present study was thus aimed to determine whether NO exerts a tonic regulatory effect on the expression of sodium transporters in the kidney. Rats were treated with NO synthase (NOS) inhibitor, NG-nitro-L-arginine methyl ester (L-NAME), for 4 weeks, and the expression of Na, K-ATPase, NHE3, BSC-1, and TSC proteins was determined in the kidney by Western blot analysis.
MATERIALS AND METHODS
Animals and renal function
Male Sprague-Dawley rats (200-250 g) were used. All procedures were done in accordance with Institutional Guidelines for Laboratory Animal Care and Use. Experimental group of rats was treated with L-NAME (100 mg/L drinking water) for 4 weeks. Control group was supplied with tap water without any drugs. Systolic blood pressure (SBP) was indirectly measured every week in a conscious state by tail-cuff method.
For the collection of urinary data, the rats were anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneal on the experimental day. The urinary bladder was exposed through a low midline abdominal incision and cannulated with PE 50 tubing to collect urine samples for 2 hr. At the end of urine collection, the arterial blood was collected. Creatinine clearance, urinary sodium excretion and fractional excretion of sodium were measured.
To determine plasma renin activity (PRA) and aldosterone level, the trunk blood was collected by the decapitation in a conscious state. For renin activity, the generation of Ang I was measured using a kit from New England Nuclear (Boston, MA, U.S.A.). For aldosterone, Coat A-Count kit was used (Diagnostic Products; Los Angeles, CA, U.S.A.).
Na,K-ATPase activity
The catalytic activity of Na, K-ATPase was determined by a coupled enzyme optical assay as described previously (13). The tissue fraction was added to the assay mixture including enzymes and other cofactors: KCl 20 mM, NaCl 130 mM, MgCl2 4 mM, ATP 3 mM, phospho [enol] pyruvate 1 mM, NADH 0.2 mM, pyruvate kinase 10 U, lactate dehydrogenase 30 U/mL and histidine 30 mM. To determine ouabain-insensitive ATPase activity, identical assay mixture added with 1 mM ouabain was used. ATP-disodium salt (5 mM), as substrate, was added to start the reaction. The absorbance was read at 340 nm at 37℃ for 2 min. The ouabain-sensitive activity was calculated by subtracting the activity measured in the presence of ouabain from the total activity. The activity was expressed as micromoles of ATP hydrolysed per mg of protein per min.
Protein preparation and Western blot analysis
The kidneys were rapidly isolated following the decapitation under a conscious state. They were rapidly frozen and kept at -70℃ until assayed. The kidney was homogenized with Polytron homogenizer at 3,000 rpm in a solution containing sucrose (250 mM), EDTA (1 mM), phenylmethylsulfonyl fluoride (0.1 mM), and potassium phosphate buffer (20 mM), at pH 7.6. The homogenate was centrifuged at 1,000 g for 15 min to remove whole cells, nuclei and mitochondria. The supernatant was then ultracentrifuged at 100,000 g for 1 hr to produce a pellet containing membrane fractions enriched for both plasma membranes and intracellular vesicles. The pellet was resuspended in homogenizing solution for protein blotting.
Protein samples (100 µg) were loaded and electrophoretically size-separated with a discontinuous system consisting of 8-12.5% polyacrylamide resolving gel and 5% polyacrylamide stacking gel. The proteins were then electrophoretically transferred to a nitrocellulose membrane at 40 V for 3 hr. The membranes were washed in Tris-based saline buffer (pH 7.4) containing 0.1% Tween-20 (TBST), blocked with 5% nonfat milk in TBST for 1 hr, and incubated with 1:2,500 dilutions of polyclonal rabbit anti α-1 and β-1 subunits of Na,K ATPase (Upstate Biotechnology; Lake Placid, NY, U.S.A.), 1:750 dilutions of polyclonal rabbit anti NHE-3 (Alpha Diagnostic; San Antonio, TX, U.S.A.), 1:100 dilutions of polyclonal rabbit anti BSC-1 and TSC (Chemicon; Temecula, CA, U.S.A.) in 2% nonfat milk/TBST for 1 hr at room temperature. The membranes were then incubated with a horseradish peroxidase-labeled goat anti-rabbit IgG (1:1,200) in 2% nonfat milk in TBST for 2 hr. The bound secondary antibody was detected by enhanced chemiluminescence (Amersham; Buckinghamshire, England). The protein levels were determined by analyzing the autoradiogram signals using the transmitter scanning videodensitometer.
RESULTS
Renal functional parameters
A steady-state hypertension was observed following the treatment with L-NAME. SBP measured on the 4th week day was 165±4 mmHg in the experimental group, while it was 122±5 mmHg in the control (n=6 each, p<0.05). Table 1 summarizes the renal functional data. Total and fractional excretion of sodium decreased significantly, while creatinine clearance and urinary flow rate remained unaltered. PRA and the plasma aldosterone level were not significantly altered.
Expression of Na,K-ATPase
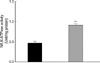
The α1-isoform specific monoclonal antibody of Na,K-ATPase recognized a band at about 110 kDa. The β1-isoform antibody recognized a band at 52 kDa. Following L-NAME treatment, the expression of α1 subunit of Na,K-ATPase was increased, although that of β1 subunit was not significantly altered (Fig. 1). Accordingly, the catalytic activity of Na,K-ATPase was increased (Fig. 2).
Expression of NHE3, BSC1 and TSC
Fig. 3, 4 show immunoblots of NHE3, BSC1 and TSC, using the membrane preparation of the whole kidney. Affinity purified anti-NHE3 antibody recognized a band at about 87 kDa. Affinity purified anti-BSC1 antibody recognized a broad band of molecular mass at 146-176 kDa. Affinity purified anti-TSC antibody recognized a broad band centered at about 165 kDa. The expression of these transporters was increased significantly following the treatment with L-NAME.
DISCUSSION
The treatment with L-NAME induced a steady-state hypertension from the first week of measurement, as in our previous study (14). Accordingly, the total and fractional urinary excretion of sodium decreased, while creatinine clearance remained unaltered. These findings support the notion that L-NAME induces a salt-sensitive hypertension associated with increased sodium balance (10-12).
It has been known that NO can inhibit sodium transport in the nephron, which may account for the natriuresis observed in vivo. For instance, sodium nitroprusside, an exogenous NO donor, significantly inhibited the activity of Na,K-ATPase in vitro (15). NO also has an inhibitory effect on both Na/H exchange and Na,K-ATPase activity in the proximal tubule (16, 17). The inhibited chloride absorption by TALH is mimicked by basolateral administration of L-arginine and is inhibited by L-NAME (18). It has been also found that NO inhibits the activity of BSC1 in TALH and MMDD1 cells (19, 20).
Following the treatment with L-NAME, the renal expression of α1 subunit of Na,K-ATPase was increased. Accordingly, the catalytic activity of Na,K-ATPase was increased. The expression of NHE3 and BSC1 also increased. The increased expression of these transporters may represent a removal of tonic inhibitory effect of NO on these transporters.
On the other hand, the sodium transport in distal nephron is activated by mineralocorticoids, in part through increasing the expression of TSC (21). However, the abundance of TSC was increased following the treatment with L-NAME, while the plasma aldosterone level remained unchanged in the present study. It is suggested that there exists a direct tonic inhibitory effect of NO on the expression of TSC.
In summary, the expression of sodium transporters, such as NHE3, BSC1, and TSC, was increased along with the expression and catalytic activity of Na,K-ATPase following a chronic blockade of NO synthesis. It is suggested that the endogenous NO plays a regulatory role in the expression of sodium transporters in the kidney with tonic inhibition. The L-NAME-induced hypertension may in part be causally related with the upregulation of these sodium transporters, culminating in increased tubular reabsorption and retention of sodium.




 XML Download
XML Download