

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mylabris, the dried body of the Chinese blister beetle (Mylabris phalerata Pallas), has been used as Chinese medicine for over 2000 yr. Its active constituent, cantharidin (CA), has anti-tumor properties and causes leukocytosis. However, it has irritant effects on the urinary organs. Norcantharidin (NCTD), the demethylated form of cantharidin (Fig. 1), is easier to be synthesized and is relatively free from side effects. NCTD inhibits the proliferation of some cancer cells (such as HL60, K562, Bel-7402, MCF-7, Colo205, HT-29, SW480) by interrupting DNA synthesis or upregulating of the CD95 receptor and CD95 ligand on the cell surface and has antitumor activity against transplanted hepatoma in mouse model. These findings suggest that NCTD is a potential antitumor agent (1-3). However, the exact mechanism responsible for the apoptotic effect is not thoroughly elucidated.
Apoptosis, or programmed cell death, is a genetically regulated, self-destructive cellular death process that is important in development, tissue remodeling, immune regulation, and many diseases (4-7). Cysteine-dependent aspartate-specific proteases (caspases) have been demonstrated to be crucial mediators in apoptotic pathway. Caspases can be divided into two groups: initiator caspases (such as caspase-8 and caspase-9) whose main function is to activate downstream caspases, and executor caspases (such as caspase-3), which mediate apoptosis by proteolysis of specific substrates including inhibitor of caspase-activated DNase (ICAD) and antiapoptotic protein, Bcl-2 (8-12). Many Bcl-2 family proteins reside the mitochondrial outer membrane. The balance between Bax and Bcl-2 (or Bcl-xL) determines the fate of cells in many apoptotic systems. Bcl-2 and Bcl-xL can be cleaved by caspase-3 and cleavage of these proteins appears to inactivate their survival function. In response to the death stimuli, the mitochrondrial membranes are permeabilized, resulting in the release of cytochrome c. In the cytosol, cytochrome c activates apoptosis by binding and activating apoptotic protease activating factor-1 (Apaf-1)-caspase-9 complex, which form an apoptosome acting as a processing/activation center for the downstream caspase-3 (13-17).
In the present study, we demonstrate that caspases activation participated in NCTD-induced apoptosis, and up-regulaton of Bax and down-regulation of Bcl-2 (or Bcl-xL) contributed to the NCTD-induced A375-S2 cell apoptosis.
MATERIALS AND METHODS
Chemical reagents
NCTD of analytical grade purity was from the Ju-nan Pharmaceutical Works (Junan, China) and dissolved in RPMI-1640 (HyClone, U.S.A.). Caspase-8 inhibitor (z-IETD-fmk) was from Enzyme Systems (CA, U.S.A.). Caspase-3 inhibitor (z-DEVD-fmk) and pan-caspase inhibitor (z-VAD-fmk) were from Calbiochem (CA, U.S.A.). Caspase-9 inhibitor (Ac-LEHD-CHO), rabbit polyclonal antibodies against ICAD, cytochrome c, Bax and Bcl-xL, mouse polyclonal antibodies against Bcl-2, horseradish peroxidase-conjugated secondary antibody (goat-anti-rabbit or goat-anti-mouse) were from Santa Cruz Biotechnology (Santa Cruz, CA). Caspase-3, -8 and -9 Apoptosis Detection Kits were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture
A375-S2, melanoma cells, were obtained from American Type Culture Collection (ATCC, #CRL, 1872, MD, U.S.A.) and were cultured in RPMI-1640 medium (HyClone, U.S.A.) supplemented with 10% heat inactivated (56℃, 30 min) fetal calf serum (Beijing Yuanheng Shengma Research Institution of Biotechnology, Beijing, China), 2 M L-glutamin (GIBCO, U.S.A.), 100 kU/L penicillin and 100 g/L streptomycin (GIBCO, U.S.A.) at 37℃ in 5% CO2.
Cell growth inhibition test
A375-S2 cells (1.0×108 cells/L) seeded in 96-well plate (NUNKTM, Roskilde, Denmark) were cultured for 24 hr, then various concentrations of NCTD (60-480 µM) were added and cultured for 12, 24, 36, 48 hr further. MTT (thiazolyl blue, Sigma, MO, U.S.A.) test were carried out to detect cell growth using an enzyme-linked immunosorbent assay plate reader (TECAN, Austria) (18). After preincubation with given concentrations of pan-caspase inhibitor (z-VAD-fmk), caspase-8 inhibitor (z-IETD-fmk), caspase-9 inhibitor (Ac-LEHD-CHO), caspase-3 inhibitor (z-DEVD-fmk) for 2 hr, 60 µM NCTD were added and cultured for further 24 hr. Growth inhibition was evaluated by MTT method. The percentage of cell growth inhibition was calculated as follows:
Relative viability (%)={1-[A492 (control)-A492 (NCTD)]}/A492 (control)×100
Nuclear damage observed by Hoechst 33258 staining
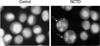
Apoptotic nuclear morphology was assessed using Hoechst 33258 (Sigma, U.S.A.) as previously described (19). A375-S2 cells, containing adherent and floating, were collected by centrifugation at 1,000 g for 5 min, washed two times with PBS. The cells were fixed with 3.7% paraformaldelyde at room temperature for 2 hr, then washed and stained with Hoechst 33258 167 µM at 37℃ for 30 min. At the end of incubation, the cells were washed and resuspended in PBS for observation of nuclear morphology using fluorescence microscope (Nikon, Osaka, Japan).
Lactate dehydrogenase (LDH) activity-based cytotoxicity assays (20, 21)
The cells were cultured with NCTD for 12, 24 or 36 hr. Floating dead cells were collected from culture medium by centrifugation (240 g for 10 min at 4℃), and the lactate dehydrogenase (LDH) content from the pellets lysed in 1% NP-40 for 15 min was used as an index of apoptotic cell death (LDHp). The released LDH in the culture medium (extracellular LDH or LDHe) was used as an index of necrotic cell death. The adherent and viable cells were lysed in 1% NP40 for 15 min to release LDH (intracellular LDH or LDHi). Then the substrate reaction buffer of LDH (L (+)-lactic acid 0.5 mM, indonitrotetrazolium 0.66 mM, phenazine methosulfate 0.28 mM, β-nicotinamide adenine dinucleotide 1.3 mM in pH 8.2 Tris-HCl) was added. The OD value at 492 nm of reaction for 1 and 5 min were assayed and LDH activities were determined by the average difference between 1 min and 5 min. The percentage of apoptotic and necrotic cell death was calculated as follows:
% apoptosis=LDHp/(LDHp+LDHe+LDHi)×100
% necrosis=LDHe/(LDHp+LDHe+LDHi)×100
Determination of DNA fragmentation by agarose gel electrophoresis
DNA extraction and electrophoresis were performed as described previously (22). In brief, A375-S2 cells, containing adherent and floating, were collected by centrifugation at 1,000 g for 5 min. The cell pellet was suspended in cell lysis buffer [Tris-HCl 10 mM (pH 7.4), EDTA 10 mM (pH 8.0), Triton-100 0.5%) and kept at 4℃ for 10 min. The lysate was centrifuged at 25,000 g for 20 min. The supernatant was incubated with RNase A 40 µg/L (Sigma) at 37℃ for 1 hr, then incubated with proteinase K 40 µg/L (Merck) at 37℃ for 1 hr. The supernatant was mixed with NaCl 0.5 M and 50% 2-propanol at -20℃ overnight, then centrifuged at 25,000 g for 15 min. After drying, DNA was dissolved in TE buffer [Tris-HCl 10 mM (pH 7.4), EDTA 1 mM (pH 8.0)] and separated by 2% agarose gel electrophoresis at 100 V for 1 hr.
Assay of caspase activities
A375-S2 cells were treated with or without NCTD. Analysis of caspase-3, caspase-8 and caspase-9 activities was performed using Caspase Apoptosis Detection Kit (Santa Cruz, CA, U.S.A.) according to the manufacturer's instruction. In brief, harvested cells at various time points were washed with PBS two times and centrifuged at 150 g for 5 min. The supernatant was aspirated off and 100 µL cell lysis buffer (provided) was added to an Eppendorf centrifuge at 500 µL per 1×106 cells. Cells in the lysis buffer were incubated on ice for 10 min. Reaction buffer containing 10 µL DTT, 10 µL of DEVD-AFC, IEVD-AFC or LEHD-AFC substrates and 380 µL H2O was added to each aliquot of cell lysate. The reaction mixtures were incubated at 37℃ for 1 hr. The fluorescence of the cleaved substrates was determined with a spectrofluorometer set at 400 nm excitation wavelength and at 505 nm emission wavelength. The unit of enzyme activity corresponds to the activity that cleaves the respective substrate in 1 min/mg protein at 37℃.
Western blot analysis
A375-S2 cells were treated with 60 µM NCTD for 0, 12, 24, 36 hr. Both adherent and floating cells were collected and frozen at -80℃. Western blot analysis was performed as previously described (23) with some modification. Briefly, A375-S2 cells were lysed for 1 hr on ice in lysis buffer [50 mM HEPES (pH 7.4), 1% Triton X-100, 2 mM sodium orthovanadate, 100 mM sodium fluoride, 1 mM EDTA, 1 mM phenylmethanesulfonyl fluoride (PMSF)], supplemented with proteinase inhibitors: 100 µg/mL aprotinin, 10 µg/mL leupeptin, and 100 µg/mL pepstatin. Protein concentration was determined by the Bio-Rad DC protein assay (Bio-Rad Laboratories, Hercules, CA). The lysate was centrifuged at 16,000 g at 4℃ for 10 min. Equivalent amounts of protein lysates were mixted in 2×loading buffer [50 mM Tris-HCl (pH 6.8), 2% SDS, 10% 2-mercaptoethanol, 10% glycerol, and 0.002% bromphenol blue], heated at 100℃ for 5 min, and then analyzed by electrophoresis in 12% SDS polyacrylamide gel and blotted onto nitrocellulose membrane (Amersham Biosciences, U.K.). After blocked with Tween 20-Tris-buffer saline [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.02% Tween 20] containing 5% nonfat milk at room temperature with the primary antibodies at 1:500 dilution in blotting buffer. After washed 3 times for 10 min each in Tris-buffered saline, the membrane was incubated with a diluted horseradish peroxidase-labeled secondary antibody (1:500) in blotting buffer at room temperature for 1 hr. After 3 more washes, proteins were detected by chemiluminescence, according to the manufacturer's instructions (Bio-Rad Laboratories, Hercules, CA).
RESULTS
Cytotoxicity of NCTD on A375-S2 cells by MTT assay
NCTD 15 to 240 µM exerted potent inhibitory effect on A375-S2 cell growth. By 24 hr after NCTD 60 µM treatment, cell death rate reached to almost 50% (Fig. 2).
NCTD-induced morphological changes and DNA fragmentation of A375-S2 cells
In control group, A375-S2 cells were round in shape and stained homogeneously. After 24 hr treatment with NCTD, blebbing nuclei and granular apoptotic bodies appeared (Fig. 3, arrows).
DNA fragmentation as a hallmark of apoptosis was observed in NCTD-treated A375-S2 cells (Fig. 4).
Apoptosis of A375-S2 cell death identified by LDH released assay
The ratio of apoptotic A375-S2 cells increased from 4.6% at 0 µM NCTD to 38.5% at 240 µM NCTD, however, that of necrotic cells were still negligible (Fig. 5).
Effect of caspases on NCTD-induced cytotoxicity in A375-S2 cells
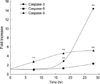
A375-S2 cells were treated with 60 µM NCTD for 24 hr in the absence or presence of various caspase inhibitors: pancaspase inhibitor (z-VAD-fmk, 40 µM), caspase-3 inhibitor (z-DEVD-fmk, 20 µM), caspase-8 inhibitor (z-IETD-fmk, 20 µM), caspase-9 inhibitor (Ac-LEHD-CHO, 20 µM). Z-VAD-fmk, z-DEVD-fmk and Ac-LEHD-CHO partially blocked NCTD-induced A375-S2 cell apoptosis. Inhibitory ratio was 30.98%, 38.96%, and 51.50%, respectively. However, caspase-8 inhibitor did not affect the death ratio (Fig. 6). Caspase-3 activity was significantly enhanced after the cells were treated with NCTD 60 µM at different time points, however, caspase-9 activity increased moderately and increase of caspase-8 activity was almost negligible (Fig. 7).
The ICAD protein degradation was significant after 24 hr incubation with NCTD (Fig. 8), and this change was blocked by caspase-3 inhibitor.
Involvement of mitochondrial proteins in NCTD-induced A375-S2 cell apoptosis
After incubation with NCTD, protein expression ratio of Bcl-xL/Bax and Bcl-2/Bax was down-regulated, and this change was blocked by caspase-3 inhibitor. At the same time, the protein of cytchrome c was increased in cytosol and caspase-3 inhibitor had no influence on the increase (Fig. 9).
DISCUSSION
The present study showed that NCTD inhibited A375-S2 cell growth in a time- and dose-dependent manner. At the same time we demonstrated that NCTD induced apoptosis in A375-S2 cells and the apoptosis was possibly associated with caspases. Caspases are a family of cysteine proteases that are activated during the apoptotic processes. Death receptors such as Fas induce caspase-8 activation via Fas-associated death domain protein (FADD). It was reported that in human colorectal carcinoma cell lines NCTD induced apoptosis by activation of caspase-8, which was prevented by the pan-caspase inhibitor z-VAD-fmk and caspase-8 inhibitor z-IETD-fmk (1), but the inhibition pathways of NCTD-induced apoptosis remain unclear. Morphological observation, DNA fragmentation and LDH activity assay suggested that NCTD induced A375-S2 cell death involved a mechanism of apoptosis. In the present study, NCTD-induced A375-S2 cell death was blocked by pan-caspase inhibitor, indicating that caspase family play a role in the apoptotic process. Caspase-3, and caspase-9 inhibitor (z-DEVD-fmk, Ac-LEHD-CHO, respectively) partially blocked NCTD-induced A375-S2 cell apoptosis, whereas caspase-8 inhibitor (z-IETD-fmk) had no effect on A375-S2 cell death induced by NCTD, indicating that post-mitochondrial caspase-9, but not upper stream caspase-8, activated this apoptotic process.
Chemotherapeutic agents and UV irradiation cause release of mitochondrial cytochrome c, which binds to Apaf-1, and this complex mediates recruitment of procaspase-9 and activates caspase-3. ICAD is expressed as two isoforms, ICADL/DFF45 and ICAD-S/DFF35. Once ICAD/DFF45 is cleaved by caspase-3 or caspase-7, CAD is released to the nucleus and induces DNA fragmentation, resulting in the morphological and biochemical features of apoptosis (9, 10, 20, 24, 25). To further confirm the participation of different caspases in the cell death, we examined the activities of caspase-3, -8, and -9 and the protein expression of cytochrome c and the substrate for caspase-3, ICAD. The activities of caspase-9 and caspase-3 were up-regulated at 8 and 18 hr, respectively, but capase-8 activity just showed slight change after 28 hr treatment. The protein expression of cytochrome c was up-regulated and the protein expression of ICAD was significantly down-regulated. Based on these results, we concluded that caspase-9-activated apoptotic pathway played a role in the apoptotic pathway of NCTD-treated A375-S2 cells. On the other hand, since NCTD-induced A375-S2 cell apoptosis was only partially reduced by caspase inhibitors, it is possible that other apoptotic pathways might also participate in this process.
Several pro-apoptotic proteins, such as Bax, Bak, and Bid, translocate to the mitochondrial membrane, and this localization is associated with their pro-apoptotic activities. It has been reported that Bcl-2 could exert its action through heterodimerization with Bax. Meanwhile, Bcl-2 and Bcl-xL are cleaved by caspase-3 and are converted to pro-apoptotic proteins similar to Bax. Therefore, the ratio between Bcl-2 and Bax or Bcl-xL and Bax is a decisive factor to activate cell death (9, 15, 26-29). A375-S2 cells treated with NCTD exhibited the elevated ratio between pro-apoptotic Bax and anti-apoptotic Bcl-2 or Bcl-xL. The oligomerization of Bax in the mitochondrial membrane has been shown to induce cytochrome c release, meanwhile pro-apoptotic Bcl-2 cleavage product was reported to localize on mitochondrial membrane and caused release of cytochrome c (9, 30, 31). In our study, the protein expression of cytochrome c was markedly up-regulated followed by the changes of Bcl-2/Bax and Bcl-xL/Bax ratios at 24 hr in A375-S2 cell treated by NCTD. At the same time, the change of Bcl-2/Bax or Bcl-xL/Bax ratio was blocked by caspase-3 inhibitor, and caspase-3 inhibitor had slight effect on the expression of cytochrome c, indicating a potential positive feedback loop that ensures the death of the cell. These results suggested that the mitochondrial pathway of cell death, including Bcl-2 family and cytochrome c, might be involved in A375-S2 cell death and orchestrate the caspase cascades.
In conclusion, NCTD inhibited A375-S2 cell growth and caspase-9, caspase-3 activation was involved in the apoptotic progression. Simultaneously mitochondrial pathway, including cytochrome c, Bax and Bcl-2 (or Bcl-xL), contributed to the NCTD-induced A375-S2 cell apoptosis. More detailed mechanism of NCTD-induced A375-S2 cell apoptosis remains to be elucidated.






 XML Download
XML Download