

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The pulmonary lymphatic system plays an important role in the lung perfusion homeostasis. Congenital errors of lymphatic vessel development lead to primary pulmonary lymphatic disorders, including lymphangiomas, lymphangiectasis, lymphangiomatosis, and lymphatic dysplasia syndromes (1). Among them, pulmonary lymphangioma is a rare benign lesion thought to result from the development of abnormally proliferating lymphatic vessels. The abnormal vessels may be capillary, cavernous or cystic in type. The basic defect has been considered to be an abnormality in the developmental lymphangiogenesis (2).
We present a case of intrapulmonary cystic lymphangioma involving the upper lobe of the right lung, which presented with dyspnea in a 2-month-old female infant, and review the literature on its pathogenesis, clinicopathologic features, and radiographic findings.
CASE REPORT
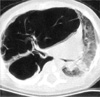
A 2-month-old Korean baby girl developed dyspnea and irritability about a week before admission to our hospital. On physical examination at admission, her temperature was 36.2℃, blood pressure 80/60 mmHg, pulse 145/min, respiratory rate 28/min, and weight 4,200 g. Breathing sound decreased in the right lung field and was accompanied by chest wall retraction. There was no edema signs in extremities. Arterial blood gas analysis result was pH 7.26-pCO2 50.3%-pO2 97.7%-O2 saturation 96.3%. Otherwise physical examination and laboratory results proved unremarkable. On birth history, delivery occurred spontaneously at 37 weeks gestation and her birth weight was 3,000 g. A chest radiograph at admission showed a solitary cystic mass, entirely occupying the upper lobe of the right lung. High-resolution computed tomography (HRCT) of the chest depicted a well-circumscribed, multiseptate, air-filled cystic lesion in the upper lobe of the right lung, mimicking the feature of type I congenital cystic adenomatoid malformation (Fig. 1).
Under the preoperative diagnosis of congenital cystic adenomatoid malformation, she underwent bilobectomy of the upper and middle lobes of the right lung. Grossly, the large part of the upper lobe was replaced by multiple cystic spaces that contained some serous fluid and varied in size from 2-3 mm to 3.5 cm. Remained lung parenchyma is hemorrhagic and revealed emphysematous change. The middle lobe just showed emphysematous change and focal hemorrhage.
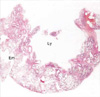
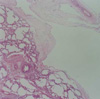
Histopathological examination of the upper lobe revealed a relatively circumscribed intrapulmonary cystic lesion composed of multiple cysts or cavities (Fig. 2). The cystic spaces were lined by a monolayer of flat or low cuboidal cells. They were interconnected each other. Their walls were supported by loose fibrous stroma with mild lymphocytic infiltrate (Fig. 3). Neither significant smooth muscle proliferation nor lymphoid follicles were observed in the walls. Lung parenchyma outside the cystic lesion exhibited emphysematous change and some lymphangiectasia in the connective tissues of interlobular septa and subpleural zones. In the immunohistochemical staining the lining cells of the cystic walls were not reactive for epithelial membrane antigen and cytokeratin, but weakly positive for factor VIII-related antigen (Fig. 4). Intrapulmonary cystic lymphangioma was subsequently diagnosed. The microscopic findings of the middle lobe were consistent with the gross finding.
The patient had an eventful recovery after operation. At 4-month follow up she remained well without evidence of recurrence.
DISCUSSION
Lymphangiomas are focal proliferation of well differentiated lymphatic tissue that presents as multicystic or sponge-like appearance. They are generally subdivided into three pathologic categories: capillary lymphangiomas describe thin-walled lymphatic channels that occur as small well circumscribed cutaneous lesions; cavernous lymphangiomas are microscopic thin-walled lymphatic channels with associated stroma; and cystic lymphangiomas are large, well-circumscribed, multiloculated cystic spaces lined by endothelium that contain a significant connective tissue component (2).
Their pathogenesis is still uncertain, but they may present as congenital or acquired forms. Congenital lymphangiomas probably represent embryologic remnants of lymphatic tissues that either failed to connect to efferent channels or arose from portions of lymph sacs that were sequestered during development (3). Acquired or secondary lymphangiomas develop in areas of chronic lymphatic obstruction related to surgery, chronic infection, or radiation.
Lymphangioma can occur in any region of the body in which there is lymphatic drainage. The single most common site of cystic occurrence is in the neck, where the lesion is referred to as a cystic hygroma. In the chest, lymphangiomas are most commonly found in the mediastinum, where they account for 0.7% to 4.5% of all mediastinal tumors (4). In a regard to intrapulmonary lymphangiomas they have been described but are extremely rare: approximately a dozen cases, with patient ages ranging from 6 months to 54 yr, are described in the English literature (1). They frequently occur in the lower lobe of the lung, which is suspected to be associated with a rich lymphatic supply noted in the part of the lung. But we also found two case reports developed in the upper lobe (5, 6), as in our case. In a review of 20 patients with thoracic lymphangioma (including mediastinal), Shaffer et al. (4) noted a slight predominance in women. But Wilson et al. (5) suggested that all adult patients with intrapulmonary lymphangioma were men, including their case. Symptoms are known to vary widely depending on the age of the patient and the extent of disease. In neonates and infants, cystic lymphangiomas often present with pneumothorax and respiratory distress (4). In our case, she complained of dyspnea developed recently, with the signs of respiratory distress. Radiologically plain radiographs, ultrasound, CT scan, and magnetic resonance imaging (MRI) have proven useful in determining the number and extent of lesions. The most common CT appearance of lymphangiomas is that of a cystic mass with smooth margins, but in the childhood the CT and/or MRI findings do not appear to be specific to differentiate lymphangiomas from other intrapulmonary cystic lesions, including congenital cystic adenomatoid malformation (CCAM), pleuropulmonary blastoma (PPB), bronchial atresia, bronchogenic cyst, and congenital lobar emphysema (7). Therefore histological and roentgenographic correlation may be necessary for establishing a definite diagnosis.
Among the list of differential diagnosis, CCAM is a disorder of embryonic bronchopulmonary development, characterized by a multicystic mass of pulmonary tissue with an abnormal proliferation of bronchial structures. There are three types: Type I consists of a large single cyst; Type II of numerous small cysts; Type III appears as a solid mass. The prognosis is variable and depends on the size and the histologic type (8). PPB is a rare malignant dysontogenetic neoplasm affecting children, and is characterized histologically by a variably mixed blastematous and sarcomatous patterns. PPB has also known to have three pathological varieties, including predominantly cystic type I, cystic and solid type II, and predominantly solid type III (9). The solid areas of the types II and III PPB are composed of undifferentiated blastemal tissue which may overlap with spindle cell sarcomatous, rhabdomyosarcomatous, anaplastic, and chondrosarcomatous foci. Bronchial atresia may be present as an asymptomatic mass in the newborn period or the lung may appear as a fluid-filled, pulmonary mass at birth. Later the fluid is replaced by air and the lobe becomes emphysematous (10). Bronchogenic cyst accounts for approximately 3% of mediastinal masses in the pediatric age group. Though the middle and posterior mediastinum are generally regarded as the commonest sites, the cysts can unusually show the intrapulmonary localization (11). When a communication persists between the tracheobronchial tree and the cyst, an air-fluid level is present as well as a history of multiple infections. A pseudostratifed, ciliated columnar epithelium, submucous glands, and isolated islands of cartilage are the composite of histologic findings for an unequivocal diagnosis of bronchogenic cyst. Congenital lobar emphysema or congenital lobar hyperinflation affects only one of the upper lobes or the right middle lobe of the lung. The main pathologic change consists of massive overdistention of the alveolar spaces, not accompanied by the destruction of the tissues. It is therefore not truly a cystic or an emphysematous process. In addition, intrapulmonary lymphangiomas need to be differentiated from other primary pulmonary lymphatic disorders, including lymphangiectasis and lymphangiomatosis (1). Lymphangiectasis is the pathologic dilation of lymphatics. Primary (congenital) and secondary forms have been described. The primary form presents in neonates and is usually fatal. Secondary forms of lymphangiectasis result from a variety of processes that impair lymph drainage and increase lymph production. Pathologically the lesion shows multifocal lymphatic dilations along the lymphatic routes of the interlobular septa and the bronchovascular bundles. But it does not show the localized proliferation of anastomosing lymphatic channels which is characteristic of lymphangiomas. Lymphangiomatosis literally describes the presence of multiple lymphangiomas. It is frequently associated with other lymphatic-related abnormalities and usually (in 75% of cases) involves multiple organs. The histology of lymphangiomatosis resembles a lymphangioma, but can appear to infiltrate tissues, raising concern for a more aggressive lesion. Clinically it presents at a later age than solitary lymphangiomas either because of an influence of hormonal factors or because a more subtle, albeit more widespread, defect requires a longer period for growth.
The natural history of intrapulmonary lymphangiomas in infants is not well-known, but surgical resection should be considered in the light of the neoplastic nature of these lesions and of the difficulty in clinically differentiating them from other cystic lesions occurring in the age class (12). If the lesion is small and localized, the application of thoracoscopic surgery is becoming increasingly popular. Incomplete resection, however, can result in recurrence at sites of surgical resection, because of their autonomous growth and also by their reaccumulation of lymph fluid (1). Prior to the surgical exploration and excision, it is prudent to investigate for other lymphangiomatous lesions and associated congenital anomalies.
In summary, we present a case of an intrapulmonary cystic lymphangioma causing dyspnea in a 2-month-old infant. To the best of our knowledge this case is the second report in the Korean literature since 1997 (13). Despite of the scarcity, this lesion should be included in the differential diagnosis of multicystic lung lesion noted during infancy.
 XML Download
XML Download