

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Previous studies have reported that the main pathological features in temporal lobe epilepsy (TLE) are Ammon's horn sclerosis (AHS) or hippocampal sclerosis (HS), neoplasia and glioneuronal malformations (1, 2). Our neuropathologic studies with 202 TLE cases have demonstrated that the most common epileptogenic cause is AHS, however, most of the cases presented the combination of AHS/HS and neuronal migration disorder (NMD) as a type of dual pathology (3).
NMD lesions, including cortical dysplasia (CD) or microdysgenesis, are caused by impeded normal neurogenesis and migration of neurons during the fetal brain development (3, 4). Cortical dysplasia (CD) is a term to describe the histopathologic changes in disorders of cerebral cortical development, such as localized or diffuse cortical abnormality, lissencephaly, agyria/pachygyria, polymicrogyria, schizencephaly, and hemimegalencephaly (4, 5). Cortical malformations are a broad category of congenital brain abnormalities caused by altered development during the formation of the cerebral cortex and are frequently epileptogenic (6, 7). Some of the NMDs clearly have a genetic etiology, however, the etiology is unknown in a large number of sporadic cases. While NMD lesions are frequently found in neocortical epilepsies, the epileptogenic mechanism is not sufficiently studied.
Animal models of NMD have been generated in rodents by intrauterine exposure to methylazoxymethanol acetate (MAM), ionizing radiation, or ethanol, and prenatal cortical freezing (8-11). In the neocortex of NMD animals induced by MAM and irradiation, there were characteristic histopathologic features similar to those found in human NMD, and epileptogenic susceptibility was increased (12, 13).
A recent study has demonstrated that NMDAR2 subunit protein is selectively expressed in the dysplastic neurons of the human epileptic neocortex (14). Rapid excitatory signal transduction is mediated by activating the postsynaptic glutamate receptors (15). One subtype of which, α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors transmit fast excitatory synaptic potentials, while another subtype, N-methyl-D-aspartate (NMDA) receptors, which have ligand-gated ion channels with voltage-dependent properties, mediate prolonged neuronal depolarization (16). The present study was undertaken to investigate the correlation of histopathologic changes and glutamate receptor abnormalities in an experimental NMD.
MATERIALS AND METHODS
Experimental NMD in rats by irradiation
Wistar rats were housed and maintained according to the animal care guide lines of the Research Institute of Medical Science, Chonnam National University. Age-matched pregnant rats, 20 weeks old, were exposed to 240 cGy of external radiography irradiation from a linear accelerator (Mevatron 7600, Siemens, U.S.A.) on gestational day 17 (E17). Gestational day 1 (E1) was counted when insemination was detected by vaginal smear. Newborn rats were obtained from the irradiated mother rats, and they were maintained carefully. Ten weeks old rats were used in this study. There were four experimental groups consisting of control (n=10) and NMD rats (n=30) for clinical, histopathological and immunohistochemical study.
Clinical and EEG monitoring
For the clinical observation of seizure development and EEG monitoring, a stereotaxic operation was carried out from control (n=5) and NMD rats (n=20). Nine weeks old rats were anesthetized with intraperitoneal injection of sodium phenobarbital (50 mg/kg), and fixed in an animal stereotaxic frame (David Kopf, Tujunga, CA, U.S.A.). Bipolar electrodes were placed in the left frontal cortex and hippocampus. Electrodes were fixed on scalp with dental cement and connected to an electrode socket. The rats were let free for a week to recover from the operation. For standard EEG recording, the electrode socket were connected via freely moving interface cables to an EEG monitor. Development of spontaneous seizure or seizure induced by an intraperitoneal injection (1 mg/kg) of kainic acid (KA, Nacalitesque, Kyoto, Japan) was monitored continuously for 5 hr in each rat. The clinical seizures were rated on 5 stages (17) with respect to gradual progression: (i) rhythmic mouth and facial twitching, (ii) head nodding and wet-dog shakes, (iii) forelimb clonus, (iv) rearing, and (v) loss of balance and falling.
Histopathology and immunohistochemistry
Ten weeks old rats, control (n=5) and NMD (n=10) groups, were anesthetized with intraperitoneal injection of sodium phenobarbital (50 mg/kg), and then they were perfused intracardially with phosphate-buffered saline (PBS, pH 7.40). The brains were removed, and fixed in buffered formalin for 48 hr. Serial coronal sections, 3 mm thick, were made from the formalin-fixed brains, and gross abnormalites were described. Then, the brain sections were embedded in paraffin, and cut with a microtome into 6 m thick tissue sections.
For histopathologic evaluation, the tissue sections were stained with hematoxylin and eosin (H&E). Bielschowsky silver stain and immunohistochemical stains for glial fibrillary acidic protein (GFAP, Dako, Glostrup, Denmark), neurofilament protein (NF-M/M, Sternberger-Mannheim, Luthervill, MD, U.S.A.), microtubule-associated protein 2 (MAP2, Boehringer-Mannheim, Germany), vimentin (Dako, Glostrup, Denmark) were performed in selected cases. For the investigation of glutamate receptor changes in human and experimental rat NMDs, a series of antibodies for glutamate receptor subunits (14, 18, 19) were used (Table 1). Tissue sections from the control and NMD brains were collected on probeglass slides (Fisher Scientific, Pittsburg, PA, U.S.A.), and processed for immunohistochemistry by avidin-biotin conjugation method (20). Tissue sections were incubated in 1.5% H2O2 in phosphate buffered saline (PBS), treated in 10% normal goat serum in PBS for 30 min in order to block charged sites on the tissue surface, and then incubated with primary antibodies throughout overnight at 4℃. After treatment with 1% avidin-biotinylated horseradish peroxidase (Research Genetics, Huntsville, AL, U.S.A.) for 1 hr at room temperature, the tissue sections were prepared for chromogen reaction with 3-amino-9-ethyl carbazole (AEC, Biomedia, Foster City, CA, U.S.A.). The sections were counter stained with hematoxylin, and examined by a light microscope.
RESULTS
Clinical data
Development of epileptic seizures and EEG abnormalities in NMD and control rats were summarized in Table 2. During the clinical observation and EEG monitoring, only one of 20 rats with NMD presented spontaneous seizures showing staring, mouth and facial twitching, and salivation. However, 17 of 20 rats had seizures after an intraperitoneal KA provocation. During behavioral seizures in NMD rats, the EEG activity was characterized by abnormally amplified rhythmic spike discharges initially developed from the left cortex and then from the left hippocampus (Fig. 1A). In the behavioral manifestations of seizures in NMD rats, 14 rats developed stage 5 seizures (orofacial twitching, wet-dog shakes, forelimb clonus, rearing and falling), and 3 rats developed stage 3 seizures (orofacial twitching, wet-dog shakes and forelimb clonus). Behavioral seizures occurred in NMD rats for 21.3±2.6 min during 5 hr period after the KA provocation, and each seizure lasted 10 to 56 sec (mean 27±6 sec). Two NMD rats showed EEG abnormality without behavioral seizure. In control groups, only one of 5 rats had seizures and the other one showed a mild EEG abnormality after an intraperitoneal KA provocation. The behavioral manifestation of seizures were staring and orofacial twitching (stage 1) in the rat. The seizure occurred for 2.3 min during 2 hr after the KA provocation, and each seizure lasted 5 to 13 sec (mean, 7±2 sec). The seizure spontaneously disappeared at 3 hr after the KA provocation.
Pathologic features
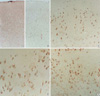
Coronal sections of the brain in control group showed well defined gray and white matter of the cerebral cortex, and intact corpus callosum. NMD group that had been irradiated on E17 consistently showed microcephaly, agenesis of corpus callosum, thinning of the gray matter of cerebral cortex, and ill-defined border of the gray and white matter (Fig. 1B). The affected cortex had nearly one half the thickness of comparable cortex in controls. Cystic changes or hydrocephalus was not noted.
Histologically, control brains showed 4 layers of the gray matter consisting of I, II/III/IV, V/VI layers of human-equivalent, and cortical subplate layer. The laminations were relatively well identified on ×40 magnification field by light microscopic examinations. In irradiated brains, histopathologic features of NMD were noted. NMD in the experimental animals was characterized by loss of normal lamination of the gray matter, neuronal heterotopia in the layer I and white matter, and presence of cytomegalic neurons. Nodular clusters of normal-sized cytomegalic neurons were seen in the layer I and subpial surface. Neurons were dyslaminated randomly throughout the gray matter of the neocotex. Nodular clusters of cytomegalic neurons in the cortex were noted in seven animals, and the lesion was more clearly identified by immunohistochemical stain for neurofilament protein (Fig. 1C). Cytomegalic neurons usually have thickened or abnormally aborizing dendrites, and some of them are abnormally oriented (Fig. 1D). Balloon cells found in some human NMD and cortical tubers were not be identified. In the hippocampi, focal disruption of pyramidal neurons in corpus ammonis with neuronal ectopia were seen in six animals. The lesions are developed at area CA1 and CA2 in 4 rats, CA2 and CA3 in 2 rats.
Immunohistochemical data for glutamate receptor subunits
Immunohistochemical expressions of glutamate receptor subunits in the control and NMD cortex are summarized in Table 3. In the control cortex, neurons in layer II/III/IV were diffusely and moderately immunoreactive to NR1 (Fig. 2A) and GluR2, and negative immunoreaction to NR2A/B and GluR2. Neurons in layer V/VI were moderately immunoreactive to NR1 and GluR3 (Fig. 2B), and mildly immunoreactive to NR2A/B and GluR2. NR2A/B presented finely granular or dot-like immunoreactivity in the neuronal cytoplasm. GluR3 immunoreactivity was limited in a few large pyramidal neurons in layer V/VI. Neurons in cortical subplate were mildly immunoreactive to NR1.
In NMD cortex, immunoreactivity for glutamate receptor subunits were demonstrated (Fig. 2C-E). Heterotopic neurons in the layer I and white matter, cytomegalic neurons, and normal-sized dysplastic neurons had strong immunoreactivity to NR1 (Fig. 2C), NR2A/B (Fig. 2D), and GluR2 (Fig. 2E). NR1 and GluR2 were labeled in neuronal cytoplasm and proximal portion of dendrites. NR2A/B showed coarsely granular immunoreactivity in dysplastic neurons. Abnormal neurons in NMD cortex were mildly immunoreactive to GluR3.
DISCUSSION
The experimental cerebral cortical malformation produced by an intrauterine irradiation was initially studied in an attempt to understand cortical development (21, 22). The present study demonstrated that the intrauterine irradiation produced NMD pathology in rats, which in turn, resulted in an increased seizure susceptibility. In this experiment, pregnant rats were exposed to 240 cGy of external irradiation from a linear accelerator on gestational day 17 (E17). Different radiation sources have been used; however, the malformative lesions were similar (10). Radiation dosages in most studies ranged from 150 to 250 cGy.
In the present study, spontaneous clinical seizures associated with epileptiform EEG activity was observed in only one out of 20 NMD rats (5%). However, most of NMD rats (90%) developed clinical seizures after small dose of KA provocation. Clinical stage, duration, and frequency of seizures were significantly increased in NMD rats than controls. These clinical data suggested that NMD might cause epileptic seizures by way of an intrinsic epileptogenecity or decreasing the threshold to excitatory neurotransmitter-induced seizures. Experimental and clinical studies suggest that NMD could increase seizure susceptibility (3, 23, 24). In particular, in vivo animal studies using an experimental model of NMD showed a decreased seizure threshold to kainic acid and to hyperthermia in the immature brain (23, 25). In vitro studies using the same experimental model suggested increased epileptiform discharges in brain tissue with experimentally induced NMD (26).
Brains from adult animals that have been irradiated on E16 or E17 show five major gross and microscopic abnormalities consisting of microcephaly, diffuse cortical dysplasia, neuronal heterotopia, focal areas of ectopic neurons in the hippocampus, and agenesis or severe hypoplasia of the corpus callosum (22, 25). Cortical dysplasia in these animals is characterized by loss of normal lamination of the neocortex, random distribution of large pyramidal cells, and nodular neuronal heterotopia. According to a pathologic grading of NMD in human (3), NMD is graded as four-layered polymicrogyria or microdysgenesis (grade I), presence of small or normal-sized dysplastic neurons with normal or loss of lamination (grade II), cytomegalic neurons (grade III), and balloon cells (grade IV). The grading of NMD rats in the present study falls in the grade III of human NMD.
In the present study, brains from 10 weeks old rats with irradiation on E17 also show gross and microscopic abnormalities. Nodular clusters of neurons in layer I up to the pial surface and the periventricular white matter are commonly seen. Focal cortical dysplasia, which is similar to grade III of NMD in human, is consistently observed in the neocortex. The focal lesion is hardly identified by gross examination, but relatively well defined by light microscopic examinations of routine H&E, or silver-stained tissue slides. Immunohistochemical stains for neurofilament proteins and MAP2 are helpful to find abnormally dense stained lesion of focal cortical dysplasia. The focal lesion is easily identified on ×2-×40 magnification field of light microscope. Cytomegalic or normal-sized neurons bearing abnormal neurofilament proteins in their cytoplasm and dendrites are frequently disoriented. Balloon cells are not noted in these animals.
Recent studies have demonstrated that NMDAR plays an important role in hyperexcitability associated with human or some animal models of human epilepsy (27, 28). Using the recently developed antibodies for glutamate receptor subunit proteins, immunohistochemical expression of NMDAR were studied in human cortical dysplasia, and increased immunoreactivities for NR1 (splice variants of NR1-1a, -1b, -2a, and -2b) and NR2A/B subunit proteins were found in resected human dysplastic cortex (14, 29-32). The immunoreactivity was localized to dysplastic neurons or cells that appear disordered compared with the normal laminar structure of cortex (14, 29). Both dysplastic neurons and nondysplastic neurons were immunoreactive to AMPA GluR2/3, but denser immunoreactivity was observed in dysplastic neurons (14). Theses reports suggest that hyperexcitability of dysplastic cortical regions possibly caused by increased expression of NR2 subunits and NR1 splice variants in dysplastic neurons.
In the present study, cerebral cortex associated NMD showed significantly increased expression of NR2A/B subunit proteins by immunohistochemistry, while control cortex showed positive reaction in a few large pyramidal neurons in the deep gray matter (layer V/VI of human cortex). NR1, GluR2, and GluR3 showed slightly increased immunoreactivities in NMD cortex than control. These results strongly suggest that NMDA receptors composed of both NR1 and NR2A/B are also increased in the NMD lesions of experimental rats. The intracellular mechanisms by which NR2A/B increases in the abnormal neurons of NMD are still unknown. NMD results from disturbances of normal development of cerebral cortex, which consists of neuroblast proliferation, neuronal migration and differentiation, and manifests in an aberrant synaptic connection, and an altered regulation of intracytoplasmic organelles (30, 33, 34). These abnormalities could lead to increased NR2A/B receptor proteins in abnormal neurons of NMD. Therefore, abnormalities in neurotransmitter and receptor, and aberrant synapses might be explained the epileptogenic mechanism of NMD.



 XML Download
XML Download