

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Pulmonary arterial hypertension progressively increases pulmonary vascular resistance, which leads to right ventricular failure and is often fatal.1) The molecular mechanisms by which endothelial vasomediators contribute to pulmonary hypertension have not been fully elucidated. Endothelin-1 (ET-1), a potent endothelium-derived vasoconstrictor peptide, was identified in 1988.2) Circulating ET-1 levels are elevated in humans who have primary and secondary pulmonary hypertension3)4) as is local pulmonary ET-1 expression,5) which suggests that this peptide may contribute to the pathogenic process. In addition, increased plasma ET-1 levels correlate with the severity of pulmonary hypertension in chronic congestive heart failure.6) ET-1 has several properties suggesting its potential pathophysiologic role in pulmonary hypertension.7) First, ET-1 contracts isolated pulmonary vessels and increases pulmonary vascular resistance. Second, ET-1 has a mitogenic effect on vascular smooth muscle cells and fibroblasts, consistent with a role in vascular remodeling, a prominent finding in pulmonary hypertension.
ET-1 responses are mediated via activation of two distinct receptor subtypes.8) ETA receptors are localized on vascular smooth muscle cells and mediate the constrictive9) and proliferative effects,10) and ETB receptors are localized on the vascular endothelium and mediate vasorelaxation11) by increasing the formation of prostacyclin and nitric oxide,12) as well as the clearance of circulating ET-1.13)14) The ETB receptors are present on the smooth muscle cells as well, where they also mediate vasoconstriction.15)
Monocrotaline (MCT) injures the endothelium of pulmonary arteries and induces progressive pulmonary hypertension in rats after a single subcutaneous (sc) injection.16) Bosentan is efficacious in experimental and clinical pulmonary hypertension.6)17-21) Bosentan improves hemodynamics, increases exercise capacity, and decreases the rate of clinical worsening in patients with World Health Organization Class III or IV pulmonary arterial hypertension.18) However, the effect of bosentan on alterations in the endothelial system and subsequent pulmonary medial hypertrophy, pulmonary hypertension, and right ventricular hypertrophy has not been established in this model.
The objectives of this study were to investigate the effect of bosentan on ET-1 and ET receptor A (ERA) gene expression in MCT-induced pulmonary hypertension.
Materials and Methods
Experimental animals
Four-week-old male Sprague-Dawley rats, weighing approximately 250 g, were used for this study. All rats were housed in climate-controlled conditions with a 12 hours light : 12 hours dark cycle, and had free access to chow and water.
Pulmonary hypertension was induced by the sc injection of 60 mg/kg MCT (Sigma Chemicals, St. Louis, MO, USA) dissolved in 0.5 N HCl solution. The rats were grouped as follows: control (n=36), sc injection of saline (0.1 mL/kg); MCT (n=36), sc injection of MCT; bosentan (n=36), sc injection of MCT plus 25 mg/kg/day bosentan (Actelion Pharmaceuticals Ltd., Allschwil, Switzerland) by gavage during all experimental days. The rats were sacrificed after 1, 5, 7, 14, 28, and 42 days. Lung tissues were removed and immediately frozen at -70℃ for enzyme analysis, post-fixed in 10% formalin, and routinely processed for paraffin embedding. All protocols were approved by the Institutional Review of Board (IRB) of the School of Medicine of Ewha Womans University.
Endothelin-1 concentration in serum
Serum was collected to determine ET-1 concentrations. The serum ET-1 concentration was measured using a human ET-1 immunoassay kit (QuantiGlo; R&D Systems, Minneapolis, MN, USA) by a sandwich enzyme immunoassay technique. A monoclonal antibody specific for ET-1 was pre-coated and immobilized onto a microplate, followed by samples with ET-1. After washing, an enzyme-linked monoclonal antibody specific for ET-1 was added to the wells. Following a wash to remove any unbound antibody-enzyme reagent, an enhanced luminol/peroxide substrate solution was added to the wells and light was produced in proportion to the amount of ET-1 bound in the initial step. A microplate luminometer was used to measure the intensity of the light emitted.
Morphometric analysis of pulmonary arteries
Hematoxylin and eosin staining was performed using 3 µm tissue sections fixed in formalin and embedded in paraffin for examination under a light microscope. The measurements of the external diameter (D) and medial thickness on either side (M1 and M2) were made along the shortest D. Measurements were made at random on 30 muscular arteries, ranging in size from 25-100 µM in D, per lung section. For each artery, the medial wall thickness was expressed as follows: % wall thickness={(M1+M2)/D}×100. In addition, the number of muscular intra-acinar arteries per field (×200) was counted. A total of 20 fields were examined in each rat for this analysis. Microscopic examination and photography were performed with a Nikon E600 microscope with a JVC digital camera attached to a phototube. The Image-pro Plus 6.0 program was used for morphometric analysis.
Ribonucleic acid extraction and complementary deoxyribonucleic acid synthesis
Total ribonucleic acid (RNA) was extracted by using TRIzol Reagent™ (Invitrogen, Carlsbad, CA, USA), according to the Trizol method protocol, and resuspended in diethyl pyrocarbonate water. Final RNA was spectrophotometrically determined at 260/280 nm. Quality was assessed as the absence of smear of 18S and 28S bands using a Bio analyzer 2100 (Agilent). RNA samples were stored at -70℃ until used. Complementary deoxyribonucleic acids (cDNAs) were synthesized by 1 µg of total RNA according to the manufacturer's protocol (High Capacity RNA-to-cDNA kit, Applied Biosystems, USA).
Gene expression analysis by real time reverse transcription-polymerase chain reaction
Real-time quantitative polymerase-chain reaction (PCR) was performed in triplicate in 384-well plates using the ABI Prism 7900 Sequence Detection System (PE Applied Biosystems) and white 384-well plates (ABgene, Hamburg, Germany) to intensify fluorescent signals. The system uses a thermal cycler and a laser that is directed via fiber optics to sample wells. The fluorescence emission from each sample was collected by a charge-coupled device-camera and the data were analyzed using the Sequence Detection System software (SDS version 2.0, PE Applied Biosystems). Reaction mixtures contained 10 pmol/µL of each primer and 2X SYBR Green PCR Master Mix (PE Applied Biosystems), which includes the HotStarTaqt DNA-Polymerase in an optimized buffer, the dNTP mix (with dUTP additive), the SYBRs Green I fluorescent dye, and ROX dye as a passive reference. Each of the PCR plates included serial dilutions (1, 1/2 and 1/4) of cDNA for generating relative standard curves.
The resulting first-strand of cDNA was normalized by the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene. The specific primers for rat ET-1 were 5'-TCTCGGAGAG CAGAGACACA-3' (forward) and 5'-TGGACTTTGGAG TTTCTCCCT-3' (reverse). The specific primers for ERA were 5'-CACAGGCTTCAGTGTGCATT-3' (forward) and 5'-CAACACAGGCCCTTAGCTTC-3' (reverse). All cDNAs were amplified using the same conditions: 50℃ for 2 minutes and 95℃ for 10 minutes followed by 40 cycles of 95℃ for 30 S and 60℃ for 30 S, then extension at 72℃ for 30 S. Melting curve analysis of products was routinely performed after amplification using an incremental temperature increase from 60℃ to 95℃ at a ramp rate of 0.21℃/sec. We then converted real-time PCR cycle numbers to gene amounts (ng).
Statistical analysis
Results were expressed as the mean±standard deviation. An unpaired two-tailed t-test and Mann-Whitney test were used, and p<0.05 was considered statistically significant. Statistical Package for the Social Sciences (SPSS) 12.0 for windows (SPSS Inc., Chicago, IL, USA) was used for all statistical analyses.
Results
Endothelin-1 concentration in serum
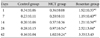
MCT significantly increased serum ET-1 concentrations from day 28 {0.97±0.54 pg/mL (MCT) vs. 0.28±0.15 pg/mL (control), p<0.05}. Bosentan increased mean serum ET-1 concentration 5-fold over MCT on day 1 {1.92±0.35 pg/mL (bosentan) vs. 0.36±0.08 pg/mL (MCT), p<0.05}, and 7 {1.19±0.40 pg/mL (bosentan) vs. 0.20±0.11 pg/mL (MCT), p<0.05}, and by 2-fold on day 14 {1.21±0.34 pg/mL (bosentan) vs. 0.57±0.36 pg/mL (MCT), p<0.05} (Table 1).
Histologic study
The basic pulmonary architecture was similar in each group. The predominant changes in pulmonary vasculature included the development of medial thickening in the pulmonary arterioles in the MCT group compared with control and bosentan group (Fig. 1). Quantitative analysis of the peripheral pulmonary arteries demonstrated that bosentan attenuated the increase wall thickness after MCT injection on day 28 and 42 (Fig. 2). Bosentan also attenuated the increased number of intra-acinar muscular arteries induced by MCT on day 1, 14, 28 (Fig. 3).
Expression of Endothelin-1 mesenger ribonucleic acid and Endothelin receptor A mesenger ribonucleic acid in rat lung tissue
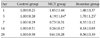
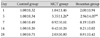
The reverse transcription-polymerase-chain reaction (RT-PCR) products from the transcripts of ET-1, ERA, and GAPDH were 156 bp, 118 bp and 89 bp (Fig. 4). MCT increased the expression of ET-1 (4.19±1.84 vs. 1±0.20) and ERA (3.35±1.26 vs. 1±0.34) genes compared with control on day 5 (p<0.05). Bosentan attenuated this increase in ET-1 on day 5 (Fig. 5) (Table 2). MCT also increased pulmonary ERA mRNA on day 5 (3.35±1.26 vs. 1±0.34, p<0.05), but bosentan did not affect this expression. There was no significant difference in the expression of pulmonary ERA mRNA among the three groups (Fig. 6) (Table 3).
Discussion
We confirmed MCT-induced pulmonary hypertension by pathologic findings and gene expression changes. The predominant changes in the pulmonary vasculature included an increased number of intra-acinar muscular arteries from 1 day and the developmental changes of medial thickening in pulmonary arteriole in MCT group from 1 week. The ratio of right ventricle to left ventricle+septum increased after 2 weeks.22) MCT also induced a significant rise in pulmonary blood pressure and apparent RVH.19)23)24) MCT also increased serum ET-1 concentrations from day 28, as well as ET1 and ERA gene expression on day 5.
The effect of endothelin receptor antagonists on MCT-induced pulmonary hypertension is controversial. Continuous infusion of BQ-12322)23) and orally active LU 135252,25) a selective ETA receptor antagonist, inhibited right ventricular hypertrophy in MCT rats. LU 135252, however, did not modify MCT-induced medial hypertrophy of pulmonary arteries. Here, bosentan reduced ET-1 gene expression on day 5 and the number of intra-acinar muscular arteries in the pulmonary arterioles from day 14. Bosentan also reduced medial thickness and RVH after 28 days, indicating a reduction in pulmonary hypertension, as shown previously.22)
We showed here for the first time that bosentan inhibited pulmonary vascular remodeling in MCT rats. Increased pulmonary artery thickness contributes to MCT-induced pulmonary hypertension and sustained irreversible increases in pulmonary vascular resistance.23) Bosentan prevented arterial medial thickening in the MCT rats, suggesting that endogenous ET-1 participates in the thickening of pulmonary vessels. Similarly, bosentan blocked the MCT-induced increase in the number of muscular intra-acinar arteries, potentially indicating neo-muscularization of non-muscular pulmonary arteries in the distal area to respiratory bronchioles. Similarly, sildenafil changed the muscularization of peripheral pulmonary arteries, with 40-50 intra-acinar arteries categorized as muscular, partially muscular, or non-muscular, although the definition of artery only included small vessels.26) Bosentan inhibited the MCT-induced increase in the number of muscular intra-acinar arteries.
MCT increased serum ET-1 concentrations on day 28. Miyauchi et al.23) reported that MCT increased plasma ET-1 levels on day 10, potentially due to differences in animal species, age, and hemodynamic characteristics. Serum ET-1 remain-ed elevated at week 4, but lung ET-1 gene expression was only elevated on day 5. Elevated serum ET-1 levels may result from increased production by the heart23)27) and kidney,23) as well as pulmonary inflammation, because the lung, kidney, and liver are the main sites for the removal of circulating ET-1.28)
Early increases in serum ET-1 putatively results from ET-1 release from storage compartments or from ET receptors. In contrast, later ET-1 elevations could result from extra-pulmonary ET-1 production, such as from ventricular myocytes responding to pressure overload. In severe pulmonary hypertension, ET-1 can be influenced by vascular surface area and receptor-mediated ET-1 clearance as well as changes in ET-1 receptor density and binding. In healthy human subjects, plasma ET-1 levels increased a maximum of two-fold within 24 hours after administration of oral bosentan.29) Loffler et al.30) found that the administration of Ro 46-2005, a bosentan analogue, increased early circulating ET-1 levels in rat plasma, suggesting that ET-1 levels are not a result of de novo synthesis.
In another MCT model, chronic intravenous infusion of a specific ETA antagonist (BQ 123) reduced pulmonary hypertension,23) whereas sc administration of another such antagonist (FR 139317) prevented RVH without affecting pulmonary hypertension.31) Bosentan (300 mg/kg/day) significantly attenuated the increase in the main pulmonary arterial pressure and RVH, and reduced plasma catecholamines and mortality.19) Different potencies and pharmacokinetic profiles may explain these various findings.
MCT increased the expression of ET1 and ERA genes on day 5, and bosentan blocked this increase. Lung ET-1 production increases in rats with pulmonary hypertension 4 weeks after aorta banding.32) Thus, changes in lung ET-1 production are not consistent in animal models of pulmonary hypertension. Severe inflammation of the lung parenchyma or severe damage of the pulmonary endothelial cells caused by MCT may increase ET-1 mRNA in the lung. Prepro ET-1 mRNA expression increases in the hearts of the MCT rats at the severe pulmonary hypertensive stage,23) potentially due to heart overload. Our data suggest that extra-pulmonary ET-1 production or trans-pulmonary ET-1 clearance might chan-ge ET-1 concentrations in serum.
In conclusion, bosentan reduced the development of MCT-induced pulmonary hypertension and subsequent RVH and vascular remodeling. These results suggest that ET-1 may play a role in RVH and remodeling in MCT-induced pulmonary hypertension. Increased ET-1 gene expression was an earlier marker than other pathologic findings or serum ET-1. Further studies are in progress to determine the effects of bosentan on other gene expressions in MCT-induced pulmonary hypertension in rats.





 XML Download
XML Download