

 PDF
PDF ePub
ePub Citation
Citation Print
Print



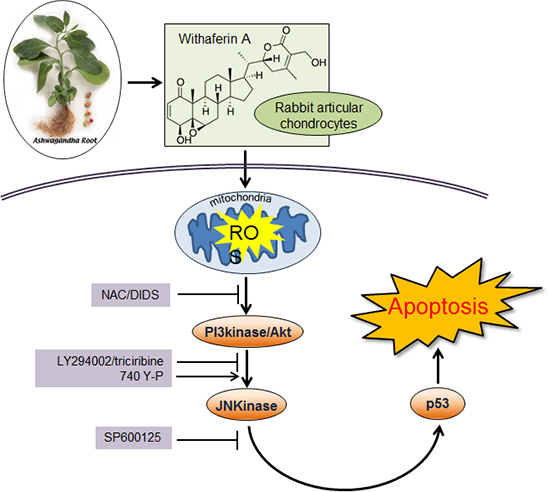
INTRODUCTION
Osteoarthritis (OA) is the most common form of arthritis related with various risk factors including age, gender, obesity, and mechanical factors, such as malalignment and abnormal joint shape and is characterized by progressive degradation of articular cartilage, and apoptosis and death of chondrocytes (1). With worldwide population ageing, the rate of OA patient is rapidly growing; and it is expected that OA will become the fourth leading cause of impairment in the coming decades (2).
Apoptosis is a critical process concerned with growth, development and homeostasis (3). Recently, the high rate of apoptosis has been observed in degenerative diseases of chondrocytes in patients suffering from OA (4). Moreover, studies by other investigations have shown that rate of apoptosis is more increased in human OA cartilage than in normal cartilage (5). Thus, the enhanced apoptosis of chondrocytes is now accepted to be a sign of progressive cartilage degradation in OA (6). Although apoptosis is recognized to have prominent role in the progression of OA, chondrocytes apoptosis and apoptosis pathways related with the pathology of OA remain largely unknown (7). Therefore, it is necessary to clarify the molecular pathways associated with chondrocytes apoptosis in OA. Over the past decade, there has been significant scientific effort for understanding of OA using a variety of mediator that contributes the cartilage damage. However, the molecular mechanisms that play a critical role in destruction of cartilage are not well understood.
The reactive oxygen species are produced in cartilage and other tissues during normal and oxidative stress. Oxidative stress carries out damage to lipids, carbohydrates, DNA and proteins, which is a process involved in degenerative diseases such as OA (8). Recent work indicates that intracellular ROS can be served as second messengers for transduction of various signals and other adaptive responses (9).
Withaferin A (WFA) is a steroidal lactone isolated from Withania somnifera. Although a variety of pharmacological activities in experimental animals has been reported (10), the molecular mechanisms underlying the function of WFA on rabbit articular chondrocytes has not been fully investigated.
The aim of the present study was to examine the effects of WFA on apoptosis of chondrocytes and to determine whether its apoptotic pathways are involved in intracellular ROS generation. Our findings indicate that WFA causes apoptosis in rabbit articular chondrocytes, which may be dependent on intracellular ROS generation.
MATERIALS AND METHODS
Reagents
Withaferin A was purchased from Calbiochem (San Diego, CA, USA), and SP600125 (SP) to inhibit JNKinase was obtained from ENZO Life science (Plymouth Meeting, PA, USA). LY294002 (LY) to inhibit PI3Kinase and 740Y-P to activate PI3K were purchased from Tocris Bioscience (Ellisville, MO, USA). Triciribine (TB) to inhibit Akt kinase was purchased from Calbiochem. N-acetyl-L-cysteine (NAC), NG-Monomethyl-L-arginine, monoacetate salt (L-NMMA) and hydrogen peroxide (H2O2) were obtained from Sigma-Aldrich (St. Louis, MO, USA). 4,4'-Diisothiocyano-2,2'-stilbenedisulfonic acid (DIDS, Sigma-Aldrich) to inhibit mitochondrial anion channel was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-p53, anti-p21 and anti-actin antibodies were from Santa Cruz Biotechnology. The anti-phospho-Akt, anti-phospho-JNK and anti-phospho- c-Jun antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). All other reagents or drugs were of analytical grade.
Culture of primary chondrocytes and experimental conditions
Rabbit articular chondrocytes were obtained from the 2-week-old New Zealand White rabbits. Cartilage slices were digested for 6 hr in 0.2% collagenase type II (381 units/mL of solid, Sigma Aldrich) in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, CA, USA) until the fragments were digested. After collecting individual cells by brief centrifugation at 300×g at 25℃ for 10 min, the cells were suspended in DMEM supplemented with heat-inactivated 10% (v/v) bovine calf serum (Invitrogen), 50 µg/mL streptomycin (Sigma-Aldrich), and 50 units/mL penicillin (Sigma-Aldrich). Released cells were seeded at 5×104 cells/cm2 in 35 mm culture dishes and incubated for 3 days at 37℃ in a humidified 5% CO2/95% air incubator for stabilization. After about 3 days in culture, chondrocytes were treated with the indicated pharmacological reagents. The study was approved by the ethics committee of Kongju National University, Gongju, Korea.
Cell viability assay
Cells were plated in a 96-well culture plates at a density of 1×104 cells/well in a final volume of 100 µL DMEM. After incubation for 3 days, the cells were treated with varying concentrations of WFA for specific time periods. After treatment, 10 µL of MTT solution (Sigma-Aldrich) dissolved in the culture medium at the final concentration of 5 mg/mL as added to each well and the plates were incubated for 4 hr at 37℃. After completing the incubation, 100 µL of solubilization buffer (10% SDS with 0.01 N HCl) was then added to solubilize MTT tetrazolium crystal, and the cells were incubated overnight at 37℃. Finally, the optical density was determined at 595 nm by using a microplate assay reader (Molecular Devices, Sunnyvale, CA, USA). The effect of WFA on cell viability was expressed as percent cell viability compared with vehicle-treated control cells, which were arbitrarily assigned 100% viability.
Cell cycle analysis
The cells were serum starved for 24 hr to synchronize them in the G0 phase of cell cycle. Synchronous populations of cells were subsequently treated with WFA for 24 hr. The cells were washed twice with cold PBS and then centrifuged. The pellet was fixed in 70% (vol/vol) ethanol for 1 hr at 4℃. The cells were washed once with PBS and resuspended in cold PI solution (50 µg/mL) containing RNase A (0.1 mg/mL) in PBS (pH 7.4) for 30 min in the dark. Flow cytometry were performed using flow cytometer (Partec GmbH, Münster, Germany). Forward light scatter characteristics were used to exclude the cell debris from the analysis. The sub-G1 population was calculated to estimate the apoptotic cell population.
Western blot analysis
For Western blotting, cells were lysed in cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, and 0.1% sodium dodecyl sulfate [SDS], supplemented with protease inhibitors and phosphatase inhibitors). Protein concentrations were determined by using a bicinchoninic acid assay (BCA) protein assay kit (Sigma Aldrich). Bovine serum albumin used as a standard. Equal amounts of total cellular proteins were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose (NC) membranes (Whatman Schleicher and Schuell, Dachen, Germany). The nitrocellulose sheet was blocked with 5% non-fat dry milk in Tris-buffered saline at room temperature for 1 hr. Antibodies were used for probing corresponding NC blots overnight at 4℃. Membranes were then washed three times with Tris-buffered saline/Tween-20 and incubated with horseradish peroxidase-conjugated secondary antibody (Sigma-Aldrich) for 2 hr. The blots were washed and then developed by use of an EZ-Western detection kit (DaeilLab service, Seoul, Korea) the protein bands were visualized using a Fuji LAS-4000 imager (Fuji Film Co., Tokyo, Japan) according to the manufacturer's instructions.
Immunofluorescence staining
Chondrocytes were plated in 35 mm culture dishes containing coverslips. After different reagents treatment, these cells were fixed with 3.5% paraformaldehyde in PBS for 15 min at room temperature and were permeabilized in PBS containing 0.1% Triton X-100 for 15 min. The fixed cells were washed with PBS and incubated for 15 min with DAPI (0.1 µg/mL, Molecular Probes, Invitrogen) at room temperature. Next, the cells were washed three times with PBS, and observed under a fluorescence microscope (BX51, Olympus, Tokyo, Japan).
Quantification of intracellular ROS production
Cells were collected and washed once with PBS and then cells were labeled with 10 µM DCFH-DA (Molecular Probes) in DMEM medium without phenol red for 30 min at 37℃ in the dark. Cells were then washed three times with PBS, and intracellular ROS levels (fluorescence intensity) were determined by flow cytometry (Partec, excitation at 495 nm and emission at 529 nm). Relative fluorescent intensities were quantified on an FLx8000 fluorescent microplate reader (Bio-Tek, VT, USA) at the indicated times. For visualization of intracellular ROS by fluorescence microscope, cells in plated in 35-mm dishes containing coverslips. Fluorescence was observed using a fluorescence microscope (BX51, Olympus).
RESULTS
WFA and induction of apoptosis in rabbit articular chondrocytes
To investigate the role of WFA in proliferation of chondrocytes, cells were treated with 1-5 µM WFA and levels of proliferation were measured using MTT assay (Fig. 1A). WFA clearly resulted in concentrations (left panel) - and time (right panel) -dependent decrease in proliferation levels (Fig. 1A). As compared with controls, levels of cell proliferation were significantly decreased by 5 µM WFA, showing 42% reductions in proliferation levels in chondrocytes (Fig. 1A). To further explain the suppression of cell proliferation by WFA, we studied apoptosis by DAPI staining (Fig. 1B) and flow cytometry analysis (Fig. 1C). The staining of cell nuclei with DAPI revealed morphological characteristics of apoptosis, including chromatin condensation and nuclear fragmentation (Fig. 1B, arrow). The nuclei in control cells showed equal distribution of the chromatin, while the apoptotic cells displayed condensed chromatin that is brightly and uniformly stained by DAPI (Fig. 1B). These morphological changes indicated that the occurrence of cell death after treated with WFA (Fig. 1B). To obtain a quantitative measure of apoptosis induction, we next examined the amount of cells with sub-G1 phase DNA content using flow cytometry analysis (Fig. 1C). Generally, the dramatic accumulation of cells in the sub-G1 phase is considered as another marker for apoptosis. WFA-treated chondrocytes resulted in significantly accumulation of cells with sub-G1 DNA content in a dose-dependent manner by 35.6%, whereas only 1.2% apoptotic cells were found in control cells (Fig. 1C).
These results suggest that WFA significantly inhibits proliferation and stimulates apoptosis (Fig. 1).
WFA and generation of intracellular ROS in rabbit articular chondrocytes
Because it is well established that apoptosis is associated with the generation of intracellular ROS (11), we examined the effects of WFA on the generation of intracellular ROS in chondrocytes (Fig. 2). Chondrocytes were treated with 5 µM WFA for 1 hr before intracellular ROS levels were detected using 10 µM DCF-DA. Hydrogen peroxide (H2O2, 200 µM) treated cells were used as a positive control (Fig. 2). Several reports have demonstrated that nitric oxide (NO) was increased in degenerative process of cartilage (12). In this regard, we also examined whether the generation of NO is involved in WFA-induced intracellular ROS production and the intensity of DCF fluorescence was determined by treatment of L-NMMA, an inhibitor of NO (Fig. 2A).
To test the hypothesis that WFA is an inducer of intracellular ROS and these effects are inhibited by NAC, a scavenger of intracellular ROS, or L-NMMA, we first examined WFA-treated cells for the accumulation of intracellular ROS detected by fluorescence microscopy (Fig. 2A, left panel). Chondrocytes were treated WFA for 1 hr and the level of intracellular ROS was monitored using DCFDA probe, which emits a green fluorescence on oxidation (Fig. 2A, left panel). The number of DCF-positive chondrocytes was caused as early as 1 hr following treatment of WFA. The peak of intracellular ROS generation was after 1 hr treatment of WFA (data not shown). A quantitative analysis showed that intracellular ROS levels were increased after the treatment of WFA by ~2-fold compared with control cells (Fig. 2A, right panel). Pretreatment of cells with NAC reduced fluorescence intensity by ~1.2-fold compared with WFA-treated cells and was returned levels to near-baseline (Fig. 2A, right panel). The results from flow cytometry analysis also showed that intracellular ROS levels were increased a significant shift of the peak (shift in relative fluorescence intensity) in cells were treated with WFA (Fig. 2B). As expected, the pre-treatment of chondrocytes with NAC left shifted the fluorescence peak closer to the peak produced by control cells (Fig. 2B). The results from flow cytometry analysis were similar to the findings from of the fluorescence microscopy (Fig. 2B). The values of DCF fluorescence were obtained at 2 min intervals up to 1 hr using a fluorometer and relative fluorescence units (RFU) values were normalized to protein content in cells (Fig. 2C). A 2.3-fold increase was observed in WFA-treated cells, whereas a decrease occurred in cells were treated with WFA in the absence of NAC (Fig. 2A, right panel and 2C). Treatment of L-NMMA was not altered the intensity of DCF-fluorescence, compared with WFA-treated cells (Fig. 2). In other words, NO did not induce WFA-caused intracellular ROS species and WFA-induced apoptosis was not related with NO generation (Fig. 2).
Our studies demonstrated that WFA causes the generation of intracellular ROS as evidenced by increase in cell population of DCF-stained cells (Fig. 2).
WFA-induced apoptosis and NAC, an intracellular ROS inhibitor
Previous studies presented that a decrease of DCF intensity was regulated by NAC but not by L-NMMA. To test the protective effect of NAC and L-NMMA on WFA-induced apoptosis, cells were treated with WFA in the presence of the indicated concentrations of NAC or L-NMMA (Fig. 3). The results indicated that WFA inhibited proliferation of chondrocytes, but these inhibitory effects were abolished by NAC (Fig. 3A). To further verify the extent of apoptosis induced by WFA, we performed PI staining to detect apoptotic cells by flow cytometry analysis (Fig. 3B). We confirmed that NAC inhibits WFA-caused apoptosis but L-NMMA does not (Fig. 3B). To confirm this, we further performed DAPI staining (Fig. 3C). Compared to the control cells, WFA-treated cells had condensed and fragmented DNA in nucleus (Fig. 3C). NAC completely blocked WFA-mediated fragmented DNA, showing that intracellular ROS production is required for the induction of apoptosis (Fig. 3). These data suggested that WFA-induced apoptosis was triggered by ROS (Fig. 3).
Effect of NAC, a scavenger of intracellular ROS, in WFA-caused intracellular ROS production
Because intracellular ROS plays a crucial role in WFA led apoptosis, it is possible that apoptotic proteins, such as p21WAF-1 and p53, which are activating under diverse stress conditions, could be changed. These proteins have an ability to mediate a DNA damage-induced apoptosis. After DNA damage, p53 activates transcription of the Cdk4 inhibitor p21WAF-1, which mediates a growth-inhibitory function and causes apoptosis (13).
Therefore, we investigated whether WFA induces apoptosis by regulating the expression of p53 and p21WAF-1 proteins. WFA-treated chondrocytes were significantly up-regulated p53 and p21WAF-1 in a dose- and time-dependent manner (Fig. 4A, B). In contrast, treatment of NAC abolished WFA-caused up-regulation of p53 and p21WAF-1 (Fig. 4C). To confirm that these cells were undergoing an apoptotic process, increasing concentrations of NAC or L-NMMA was added to WFA treated cells; and expression levels of p53 and p21WAF-1 were analyzed by Western blot (Fig. 4D, E). As a result, an induction in the protein levels of p53 and p21WAF-1 was detected in WFA-treated cells, while treatment of NAC completely inhibited their effect in concentrations-dependent manner (Fig. 4D). However, treatment of L-NMMA did not have any effect on WFA-induced the expression of p53 and p21WAF-1 (Fig. 4E). Our results clearly demonstrated that WFA is able to increase p21WAF-1 and p53, and pretreatment of NAC led to a marked inhibition of p53 and p21WAF-1 (Fig. 4). These results suggested that WFA-caused apoptosis was mediated by an increase of p53 and p21WAF-1 and their effects were prevented by inhibiting oxidative stress, ROS, but not NO (Fig. 4).
WFA and PI3K and JNKinase pathways
It has been reported that the activation of PI3-kinase (PI3K)/Akt and c-jnu N-terminal kinase (JNK)/c-jun pathways regulates cell proliferation and apoptosis in response to various stimuli (14). Therefore, PI3K/Akt and JNK/c-jun kinase pathways were examined to determine if apoptotic effect of WFA is dependent on the PI3K/Akt and JNK/c-jun pathways. As shown in Fig. 5, the phosphorylation of Akt, JNK, and c-jun increased after treatment of WFA (Fig. 5A). Levels of Akt phosphorylation began to increase at 30 min, to maximum levels at 6 hr and slightly decreased thereafter (Fig. 5B). WFA was also found to increase the activation of JNK and after that, phosphorylation of c-jun was induced till 6 hr. The activation effects of WFA on Akt, JNK, and c-jun were totally decreased or delayed by pre-treating the cells with 5 mM NAC for 1 hr (Fig. 5C). These results indicated that WFA activates Akt, JNK, and c-jun; and their activation was blocked by treatment of NAC. These findings suggest that WFA-induced apoptosis could be mediated through the alteration of activity of Akt and JNK (Fig. 5).
Apoptosis by WFA-caused intracellular ROS and PI3K/Akt and JNK/c-jun kinase pathways
To provide further evidence for the critical role of PI3K/Akt and JNK/c-jun in the apoptotic effect of WFA in chondrocytes, the PI3K-specific inhibitor LY294002, the Akt-direct inhibitor triciribine, the JNK-specific inhibitor SP600125, and the PI3-kinase activator 740Y-P were used (Fig. 6). Chondrocytes were treated with the specific inhibitor of JNK, SP600125, or with the specific inhibitor of PI3K/Akt, LY294002/triciribine for 1 hr (Fig. 6A-C). Inhibition of JNK and PI3K/Akt with SP600125 or LY294002/triciribine were abolished the expression of p53 and p21WAF-1, and inhibition of PI3K/Akt with LY294002/triciribine were prevented the phosphorylation at JNK and c-jun (Fig. 6A-C). The activation of JNK and c-jun was reduced by pre-treating LY294002 or triciribine, but the phosphorylation of Akt was not changed by treatment of SP600125 (Fig. 6A-C). The observed findings indicate that JNK/c-jun and PI3K/Akt pathways are a crucial role in WFA-caused apoptosis and the activation of JNK is a downstream of PI3K/Akt in WFA-induced apoptosis (Fig. 6). To gain further insight into the mechanism of PI3K/Akt activated by WFA, we investigated whether 740Y-P, a cell-permeable phospho-peptide capable of binding to the p85 regulatory subunit of PI3K to stimulate enzyme activity, could block the expression of p53 and p21WAF-1. Pretreatment of chondrocytes with different doses of 740Y-P, followed by 24 hr exposure to WFA continued to maintain p53 and p21WAF-1 and an increase of phosphorylation in Akt, JNK, and c-jun, suggesting the Akt pathway involved in WFA-induced apoptosis (Fig. 6D). These findings suggest that PI3K/Akt and JNK/c-jun play a crucial role in WFA induces apoptosis and also intracellular ROS caused by WFA are sufficient to activate PI3K/Akt and JNKinase pathways (Fig. 6).
Role of PI3K and JNK/c-jun kinase pathways in WFA-induced apoptosis
To establish the effects of blockage of PI3K and JNK/c-jun kinase pathways on WFA-caused apoptosis, chondrocytes were treated with SP600125 and LY294002 in the presence or absence of WFA (Fig. 7) and performed MTT assay, flow cytometry analysis, and DAPI staining. As shown in Fig. 7, by treatment of SP600125 or LY294002, decreased WFA-caused cell apoptosis and a diminished sub-G1 population recovered (Fig. 7A, B). In consistent with the result of flow cytometry analysis, nuclear morphology and DNA fragmentation of chondrocytes were reversed by treating inhibitors, as determined by DAPI staining (Fig. 7C).
Next, we confirmed that the ROS production was regulated by PI3K and JNK/c-jun kinase pathways (Fig. 7D). Inhibition of PI3K and JNK/c-jun kinase with LY294002 and SP600125 did not alter WFA-caused intracellular ROS generation (Fig. 7D). These results indicated that PI3K and JNK/c-jun kinase pathways are important for the regulating WFA-induced apoptosis (Fig. 7).
Blocking of WFA-caused apoptosis by DIDS
We previously found that intracellular ROS are generated by treatment of WFA in chondrocytes. Because the production of these intracellular ROS was diminished by specific inhibitors of mitochondrial electron transport, it was concluded that mitochondria must have been involved in their generation (15).
To clarify whether mitochondria-generated intracellular ROS regulates WFA-caused apoptosis, chondrocytes were treated with 4,4'-diisothiocyano-2,2'-stilbenedisulfonic acid (DIDS), to block anion channels in the mitochondria inner membrane, for 1 hr to inhibit the formation of intracellular ROS from mitochondria, and then treated with WFA (Fig. 8). DIDS prevented the expression of p53 and p21 in response to WFA (Fig. 8A). In WFA-treated cells DIDS also was recovered cell proliferation and prevented apoptosis (Fig. 8B). To establish if this intracellular ROS generation is sufficient to cause apoptosis, WFA-treated cells were analyzed by FACS and DNA fragmentation to determine of cell death (Fig. 8C, D). DIDS resulted in a decrease of apoptosis to ~2.3% in WFA-treated cells (Fig. 8C) and was recovered a fragmented DNA (Fig. 8D). These data indicate that DIDS do inhibit the ability of intracellular ROS to induce apoptosis, and are consistent with the conclusion that intracellular ROS may act as the signal upstream from mitochondria.
DISCUSSION
This is the first report demonstrating apoptotic effect of withaferin A in normal chondrocytes with possible mechanism of action. Because WFA is known as one of the most pro-apoptotic compound for a variety of cancer cells, it is suitable for study of chondrocytes apoptosis.
An increasing of chondrocyte apoptosis is considered a key pathological feature of OA and resent reports supported that OA chondrocytes appear mitochondrial dysfunction, which precedes the typical sign of apoptosis. Apoptosis of chondrocytes is a fundamental feature in OA cartilage degradation, and blockage of the pro-apoptotic pathway suppressed progression of OA (16).
To that end, the field of apoptosis related research has been moving ford at frighteningly rapid rate. Although many of the fundamental apoptotic proteins have been documented, the molecular mechanisms and interaction of the discovered proteins remain to be elucidated.
Increases in oxidative stress such as production of reactive oxygen species (intracellular ROS) are associated with many pathological processes (17). These intracellular ROS can act as critical signaling molecules under physiological and pathophysiological conditions (18).
Intracellular ROS mediates destroy of the extracellular matrix by causing of chondrocytes apoptosis in cartilage (19). There is increasing evidence that oxidative stress such as intracellular ROS regulates apoptosis and cell cycle arrest in a wide variety cells.
We investigated the effect of WFA on apoptosis of rabbit articular chondrocytes with a focus on ROS production. The principle finding of this investigation is that an essential mechanism for the destroyed actions of pro-apoptotic regents on chondrocytes.
We used rabbit articular chondrocytes to determine the effect of WFA on apoptosis. We found that WFA treatment inhibits proliferation (Fig. 1A) and causes apoptosis (Fig. 1B, C) in a dose dependent-manner by MTT assay. The shift to Sub-G1 cell cycle accumulation is accompanied by a depletion of cells in the G0/G1 phase, whereas proportion in cells was largely unchanged (Fig. 1C). These result showed that WFA causes apoptosis.
ROS mediates degradation of the extracellular matrix by causing chondrocytes apoptosis in cartilage (20). We also show that intracellular ROS was specially evaluated in WFA-treated cells (Fig. 2). These results suggest that the WFA is probably able to damage leading to intracellular ROS production. In addition to the activation of signaling pathway involved in cell proliferation and apoptosis, intracellular ROS may regulate the activity of transcription factors, including p53 and p21.
To elucidate the link between apoptosis and intracellular ROS generation, we investigated the expression of p53 and p21 in cells after treatment with WFA (Fig. 4). Western blot analysis revealed a high expression of p53 and p21 in WFA-treated chondrocytes that was down-regulated after treatment of NAC (Fig. 4C).
Nitric oxide (NO), the main intracellular ROS produced by chondrocytes and NO donor, sodium nitroprussade (SNP), is able to stimulate apoptosis (21). So, we analyzed the production of NO in WFA-treated cells and confirmed that WFA did not show any affect on NO production (data not shown).
Three major mitogen-activated protein kinases (MAPKs), c-Jun N-terminal kinase (JNK), p38 and extracellular signal-regulated kinase (ERK), have been shown to modulate apoptosis (22). Production of intracellular ROS results in the activation of several kinases including MAPKs, and it shows that MAP kinases are motivated in the early stages of agent treatment. Their findings have suggested that intracellular ROS is an upstream of MAPK activation (23).
Generally, the ERK pathway is crucial role for cell proliferation and is stimulated by growth factors (24). In opposition, the JNK and p38 kinase cascades are activated by genotoxic agents and cytokines regulating the stress response, growth arrest and apoptosis (25). JNK is generally considered the kinase that phosphorylates c-Jun, which is believed to mediate death of cells in a variety of circumstance (26). The transcription factor c-Jun is a major stress-activated protein and is likely to coordinate transcriptional program in response to stress.
MAPKs in apoptosis induced by WFA were investigated using SB203580, a specific inhibitor of p38, a specific inhibitor of MKK1 and MKK2 (ERK upstream kinases), or SP600125, a specific inhibitor of JNK. The results have suggested that apoptosis were inhibited by inhibiting JNK (Fig. 6A), while did not alter by preventing p38 and ERK-1/-2 (data not shown). These results have showed that JNK is important in apoptosis of WFA-treated chondrocytes.
Phosphatidylinositol 3-kinase (PI3K) is activated via phosphorylation of Ser-473/474 and Thr-308/309, and positively and negatively regulates cell growth and survival (27). Akt is a downstream target of the PI3K signaling pathway and increasing findings have reported that oxidative stress can modulate Akt activation (28).
General findings suggested that the activation of PI3K/Akt pathway has been observed in survival conditions and the overexpressed Akt can prevent apoptosis (29). However, recent accumulating reports demonstrate that the activation of PI3K/Akt occurs during apoptosis and acts as a "brake" on the process (30). These previous findings have supported the view that PI3K/Akt signaling pathway is the negative regulator for apoptosis of chondrocytes. Here, we showed that WFA could modulate apoptosis of WFA-stimulated chondrocytes through triggering PI3K/Akt pathway (Fig. 6).
To investigate the involvement of PI3K/Akt in the regulation of apoptosis by WFA-induced intracellular ROS production, the specific PI3Kinase inhibitor, LY294002, and the Akt inhibitor, triciribine, were used (Fig. 6B, C). The inhibition of PI3K/Akt showed a blockage of apoptosis, indicating that PI3K/Akt pathway is involved in WFA-induced apoptosis. Treatment of Akt activator, 740Y-P which is a cell-permeable p85 binding peptide, led to an increase in expression of p53 and p21 (Fig. 6D).
In addition, the step of ROS production by WFA is upstream of PI3K/Akt and JNK/c-Jun pathways because ROS production by WFA is apparently not influenced by the inhibitors (Fig. 7D).
In conclusion, it was shown that WFA regulates apoptosis via ROS generation. WFA-caused ROS generation activates PI3K/Akt and JNK pathways and its pathways might be important for regulating apoptosis of chondrocytes. Understanding the precise molecular pathways of the inhibition of proliferation and the induction of apoptosis in chondrocytes might lead to the search for drug screening and therapeutic strategy of OA.
Therefore, this result may contribute to establishing the scientific data basis of degenerative process for arthritis and additionally the exact mechanisms associated with oxidative stress and cartilage destruction need to be further explored.








 XML Download
XML Download