

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
The mechanisms involved in chronic myeloid leukemia (CML) progression from the chronic phase (CP) to an accelerated phase (AP) or blast crisis (BC) remain largely undetermined, but generally involve additional molecular changes besides the Philadelphia chromosome [1]. Evidence suggests that secondary BCR (breakpoint cluster region)-ABL1 (Abelson murine leukemia viral oncogene homolog 1)-dependent/independent genetic events play a critical role in blast transformation [2]. Besides BCR-ABL1 point mutations, deletions and/or rearrangements of other cancer-related genes have been associated with CML-BC [2]. Recent studies have demonstrated that aberrant activation-induced cytidine deaminase (AID) expression promotes genetic instability, eventually leading to progression and drug resistance in CML [3].
We report a patient with a rare chromosomal translocation 46,XY,t(5;12)(p13;p13) that disrupts AID and causes a concomitant deletion of the tumor suppressor gene BCL7A (B-cell CLL/lymphoma 7A). This report presents evidence of a mechanism underlying CML progression and tyrosine kinase inhibitor (TKI) resistance occurring through an interaction between AID expression and BCL7A loss in a BCR-ABL1 kinase domain mutation-independent manner.
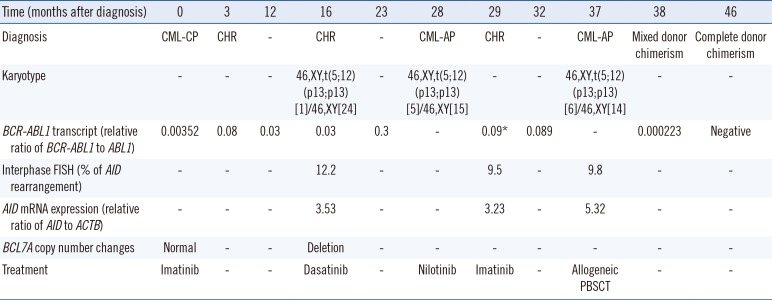
A 57-yr-old man presented in December 2010 with generalized weakness and chest pain. Bone marrow (BM) demonstrated nearly packed marrow owing to myeloid and megakaryocyte hyperplasia (blasts [0.4%]). Written informed consent for genetic analysis was obtained from the patient according to the ethical guidance of the institutional review board. The patient exhibited the b3a2 type BCR-ABL1 fusion transcript, and cytogenetic analysis revealed a normal karyotype. The patient was diagnosed as having CML-CP and treated with imatinib (Table 1).
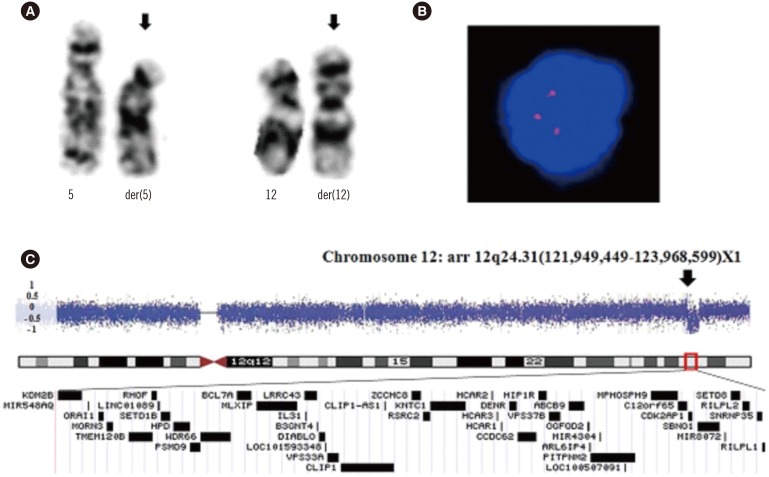
Sixteen months following diagnosis, treatment was replaced by dasatinib since BCR-ABL1 transcripts had not declined. Simultaneously, one of 25 metaphase cells exhibited an abnormal karyotype of 46,XY,t(5;12)(p13;p13) (Fig. 1A). FISH analysis using a TEL/AML1 translocation probe presented a normal result; therefore, another gene on chromosome 12p13 was suspected to be involved. Subsequent FISH analysis revealed a break in the AID signal on one allele in 12.2% of interphase cells using a custom-labeled probe (Empire Genomics LLC, Buffalo, NY, USA) (Fig. 1B) compared with 0.2% of normal control cells. Quantitative analysis showed that AID RNA expression increased, whereas normal cells showed no AID expression.
Chromosomal microarray using a CytoScan 750K Array (Affymetrix Inc., Santa Clara, CA, USA) revealed a 1.9-Mb deletion mostly containing BCL7A on chromosome 12q24.31 (Fig. 1C).
After 28 months, the patient progressed to CML-AP, and the CBC showed leukocyte counts of 116.8×109/L and 13% blasts. BCR-ABL1 kinase domain sequencing revealed no mutation. Conversely, chromosomal abnormality of t(5;12)(p13;p13) exhibited clonal expansion. Thus, the patient was returned to imatinib therapy and subsequently demonstrated a complete hematologic response; however, AID breakage and BCR-ABL1 transcript levels remained detectable.
After 37 months, the patient presented again with CML-AP, with 13.5% blasts in peripheral blood, and karyotyping showed 46,XY,t(5;12)(p13;p13)[6]/46,XY[14]. The patient received allogeneic stem cell transplantation and achieved complete donor hematopoietic chimerism.
The AID gene, located on chromosome 12p13, is associated with genomic instability and ensuing oncogenesis in various human malignancies including CML, AML, and ALL [345]. AID has several non-immunoglobulin targets, including PAX5 and BCL7A [36]; PAX5 can induce TKI resistance and CML progression via AID upregulation [3].
Recently, it was shown that AID activation can induce mutations and translocations at the BCL7A locus [6]. BCL7A belongs to the BCL7 family, which includes BCL7B and BCL7C, and is a known pro-apoptotic tumor suppressor in B-cell oncogenesis. Specifically, BCL7B suppresses β-catenin expression, causing Wnt signaling inhibition [7]. Importantly, β-catenin deregulation in Wnt signaling is associated with genomic instability that can induce oncogenic translocations in T-cell lymphoma and AML [58]. In CML, β-catenin activation is observed in the AP or BC, and is believed to promote leukemic stem cell generation that facilitates disease progression [9].
We suggest a novel pathway for disease progression and drug resistance in CML via AID expression and the concomitant loss of BCL7A. Our case supports the hypothesis that BCL7A deletion induces abnormal Wnt/β-catenin signaling, enhancing the potential for a chromosomal aberration and its persistence in our patient [510]. Loss of the tumor suppressor BCL7A may cooperate with AID activity in disease progression by causing aberrant cell cycle regulation [1]. Moreover, AID-induced DNA damage checkpoint activation causes initiation of tumor suppressor genes involved in DNA repair or cell cycle control [1]. A previous study showed that decreased tumor suppressor gene expression can cause AID-induced translocation, eventually leading to cancer [10].
In conclusion, our report identifies that AID, in combination with BCL7A, likely confers TKI resistance in a BCR-ABL1 kinase domain mutation-independent manner and may be a putative functional target in CML therapy.
 XML Download
XML Download