

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Overactive bladder (OAB) has a significant negative impact on all measures of quality of life.[1] More than 40% of patients with OAB have urge incontinence, which is defined as loss of bladder control; although OAB is not a life-threatening condition, it significantly decreases quality of life.[2] OAB affects approximately 7–27% of men and 9–43% of women and becomes more common with age.[1] In 2007, the total national cost of OAB in the US was estimated at 65.9 billion US dollars, including 14.6 billion US dollars due to lost productivity.[3] Hence, new treatments such as solifenacin have been developed to care for this condition.
Solifenacin is a competitive cholinergic receptor antagonist that is selective for the M3 receptor subtype.[4] The binding of acetylcholine to these receptors, particularly M3, plays a critical role in the contraction of smooth muscle.[5] By preventing the binding of acetylcholine to these receptors, solifenacin reduces smooth muscle tone in the bladder, allowing the bladder to retain larger volumes of urine and reducing the number of micturition, urgency and incontinence episodes.[5] Because of its long elimination half-life, a once-daily dose of solifenacin can offer 24-hour control of urinary bladder smooth muscle tone.[5]
According to the VESIcare® label, peak plasma solifenacin concentrations were reached 3–8 hours after single oral administration of a 5- or 10-mg solifenacin tablet.[6] Studies in healthy adults have shown that the drug has a high absolute bioavailability of approximately 90%, which does not decrease with concomitant food intake.[67] Solifenacin is eliminated mainly through the hepatic metabolism via cytochrome P450 (CYP) 3A4, with only approximately 3 to 13% of the dose excreted unchanged in the urine, and solifenacin metabolites are unlikely to contribute to clinical effects of solifenacin.[6] The terminal elimination half-life ranges from 33 to 85 hours, permitting once-daily administration.[8] Urinary excretion plays a minor role in the elimination of solifenacin.[8]
The objective of this study was to compare the pharmacokinetics of the newly developed solifenacin tartrate, a different salt formulation of solifenacin compared to the reference formulation, solifenacin succinate (VESIcare®). In this study, the single-dose pharmacokinetic properties of the test formulation (solifenacin tartrate 10.66 mg) and reference formulation (solifenacin succinate 10 mg) were compared in healthy male volunteers.
Methods
This study was a clinical trial conducted at the Chonbuk National University Hospital clinical trial center. This study was approved by the Ministry of Food and Drug Safety (MFDS) and the Institutional Review Board (IRB No. 2016-01-023) of Chonbuk National University Hospital (Jeonju, Republic of Korea) and was conducted according to the Ethical Principles for Medical Research Involving Human Subjects outlined in the Declaration of Helsinki and the Guidelines for Good Clinical Practice. This study was registered at ClinicalTrial.gov (https://clinicaltrials.gov/ct2/show/NCT02940314).
Subjects
Healthy male volunteers (19 to 45 years old with a body mass index (BMI) of 17.5 to 30.5 kg/m2) who had no congenital abnormalities and no chronic disease within the past 3 years were recruited. These volunteers were informed about the details of the study (including the purpose, benefits, and risks) and provided signed informed consent before participating in the study. Physical examinations, measurements of vital signs, 12-lead electrocardiograms (ECGs), and clinical laboratory assessments (i.e., hematology, biochemistry, urinalysis and urine drug screening) were performed to confirm the health of the individuals within 3 weeks before the first administration of the investigational products. Individuals who had no clinically significant findings in these screening tests were eligible to become study subjects. The exclusion criteria consisted of any evidence or history of the following: hemorrhagic, renal, endocrine, pulmonary, gastrointestinal, urinary, cardiovascular, hepatic, psychiatric, neurological, or allergic disease; hypersensitivity reaction to the components of the investigational products; use of any drug known as a significant inducer or inhibitor of drug-metabolizing enzymes within 30 days before the beginning of the study; or a genetic problem such as galactose intolerance, Lapp lactase deficiency and glucose-galactose malabsorption. Subjects were asked to avoid smoking and the consumption of caffeinated food and beverages during the hospitalization periods of the study. Subject compliance with these restrictions was determined by self-reporting and by taking the subject's history.
According to previous pharmacokinetic studies, the intrasubject coefficient of variation (CVintra) of the area under the curve up to the last sampling time (AUClast) and the maximum plasma concentration (Cmax) of solifenacin were 11.8% and 11.4%, respectively.[4] Considering the increase in the coefficient of variation due to different salt formulation, the CVintra was set to approximately 20% in this study. When the true ratio (test/reference) of the mean is 1.05 and the CVintra is 0.2, a sample size of 24 achieves 90% power at a 5% significance level. The total sample size was set to 36, considering a 30% dropout rate.
Study design
This study was a clinical trial conducted at the Chonbuk National University Hospital clinical trial center. The study was designed as a randomized, open-label, single-dose, two-way crossover study. Subjects were hospitalized in the clinical trial center on the evening before drug administration and were allocated to two groups in a 1:1 ratio according to a predesigned randomization table. During the first treatment period, subjects received one of the two treatments (either the test formulation or the reference formulation). The test formulation was solifenacin tartrate 10.66 mg (Besigum Tab, Hanmi Pharmaceuticals Co. Ltd., Seoul, Republic of Korea),[9] and the reference formulation was solifenacin succinate 10 mg (VESIcare, ASTELLAS PHARMR KOREA Co., Ltd., Seoul, Korea).[6] After the scheduled procedures in the first treatment period were finished (day 2), the subjects were discharged. After a 21-day washout period (more than 5 times the half-life of solifenacin), each subject received the other formulation.
After 10 hours of overnight fasting, the investigational products were administered to each subject with 150 mL of water. An oral check was immediately performed after administration to ensure compliance. All subjects received standardized lunch and dinner at 4 hours and 10 hours post-dose, respectively. Water was not permitted for 1 hour before and 1 hour after drug administration. In addition, subjects were not allowed to consume grapefruit or grapefruit-containing products from 7 days before the first drug administration until the collection of the final blood sample for pharmacokinetic assessments.
For pharmacokinetic analysis, blood samples containing 6 mL of blood each were collected to measure solifenacin concentrations during each treatment period at the following time points: pre-dose and 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 48 and 72 hours post-dose. The blood samples were immediately centrifuged at 3,000 rpm for 10 minutes at 4℃, and the plasma was transferred to polypropylene tubes and then stored in a freezer at −70℃ until further analysis.
Analytical procedures and pharmacokinetic assessments
Plasma concentrations of solifenacin were determined by a validated ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS, Waters ACQUITY UPLC™ System and Waters Xevo™ TQ MS; Waters, MA, USA) method using solifenacin-d5 as the internal standard. Chromatographic separation of the compounds was accomplished using a Waters ACQUITY UPLC®BEH C18 column (2.1 mm ID × 50 mm L, 1.7 µm; Waters, MA, USA). A gradient elution procedure was employed using 0.1% (v/v) formic acid in distilled water and 0.1% (v/v) formic acid in acetonitrile at a flow rate of 0.3 mL/min. Briefly, 10 µL of the internal standard (0.1 µg/mL) and 300 µL of acetonitrile were added to 100 µL of plasma sample. This sample was stirred for 3 min at 1,500 rpm using a MixMate (Eppendorf, Hamburg, Germany), followed by centrifugation for 1 min at 4,000 rpm. A 150-µL aliquot of supernatant was placed in a 1.1-mL deep-well plate (Axygen, NY, USA), and 300 µL of 0.1% (v/v) formic acid in distilled water was added. After stirring for 3 min at 1,500 rpm and centrifuging for 1 min at 4,000 rpm, 10 µL of diluted sample was injected into the UPLC-MS/MS system. Electrospray ionization in positive ion mode was used for detection and quantification. The multiple reaction monitoring (MRM) transitions were m/z 363.21→110.05 for solifenacin and 368.19→110.05 for the internal standard. A calibration curve covering the range of 0.200 to 100 ng/mL was constructed. For solifenacin, the intraday accuracy was 97.2% to 100.0% (with a precision of 1.0%–1.5%), and the interday accuracy was 96.3% to 100.5% (with a precision of 1.6%–6.0%). These results indicated that the bioanalytical method for the drug assay was adequate (r2≥0.9950).
Individual pharmacokinetic parameters were analyzed using Phoenix® WinNonlin® version 6.3 software (Pharsight Corporation, CA, USA) with the noncompartmental method. Of the pharmacokinetic parameters, AUClast and Cmax were evaluated as primary parameters. The area under the plasma concentration-time curve up to infinity (AUCinf), the time to Cmax (Tmax) and the plasma elimination half-life (t1/2) were also estimated as secondary parameters. AUClast was calculated using the linear trapezoidal method. AUCinf was calculated with the following equation: AUClast + Clast/λz, where Clast is the last measured concentration and λz is the elimination rate constant estimated from the log-linear terminal phase of the concentration-time curve as the slope of the natural logarithm of concentration against time. Cmax and Tmax were determined by observing the data of the plasma concentration-time profile, and t1/2 was calculated as ln 2/λz. In pharmacokinetic analyses, actual sample times were used.
Safety assessments
Safety was assessed by monitoring adverse events (AEs), physical examinations, laboratory tests (hematology, biochemistry and urinalysis), vital sign measurements (blood pressure, pulse rate and body temperature), and ECG. AEs were identified by asking the subjects about their condition during the study period. AE data were recorded from the pre-dose until the post-study visit. AEs were summarized by treatment groups in terms of severity (mild, moderate, or severe) and relationship. Physical examinations, laboratory tests, and vital sign measurements were conducted at screening, pre-dose, 72 hours post-dose, and at the post-study visit. ECGs were recorded at screening and the post-study visit.
Statistical analyses
Subjects who completed the pharmacokinetic blood sampling as scheduled were included in the pharmacokinetic analysis. Statistical analyses were performed using SAS software, version 9.3 (SAS institute, Cary, NC, USA). To summarize the pharmacokinetic data from the two treatments, descriptive statistics that included arithmetic means, standard deviations (SDs), and median values for continuous data were used. The log-transformed pharmacokinetic parameters of AUClast and Cmax were analyzed using a mixed-effects analysis of variance (ANOVA) model with fixed effects of sequence, period, and formulation and a random effect of subjects within the sequence to compare the pharmacokinetic parameters of the two formulations. The 90% confidence intervals (CIs) of the geometric least-square mean ratios of the test to reference formulations for AUClast and Cmax of solifenacin were calculated to assess bioequivalence.
Results
Subjects
Of the 45 volunteers who were screened, 36 subjects were enrolled in the study and randomized into the two treatment groups. The demographics of the subjects (mean ± SD) included a mean age of 22.1 ± 2.3 years, height of 174.7 ± 6.1 cm, weight of 69.6 ± 9.4 kg, and BMI of 22.8 ± 2.6 kg/m2 (Table 1). There were no significant differences in demographic characteristics between the sequence groups.
A total of 36 subjects were randomized in this study. Three subjects withdrew consent after the first dose and were dropped from the study. After the second period, one subject withdrew consent and did not participate in the post-study visit; however, this subject completed the scheduled pharmacokinetic sampling schedule and was included in the pharmacokinetic analysis. The pharmacokinetic assessment was conducted based on 33 subjects who completed the entire pharmacokinetic blood sampling schedule without significant violations.
Pharmacokinetic analysis
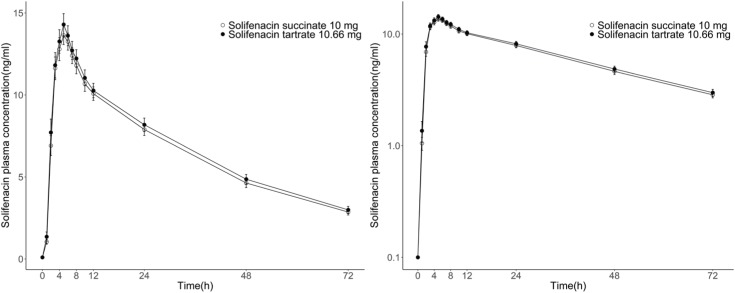
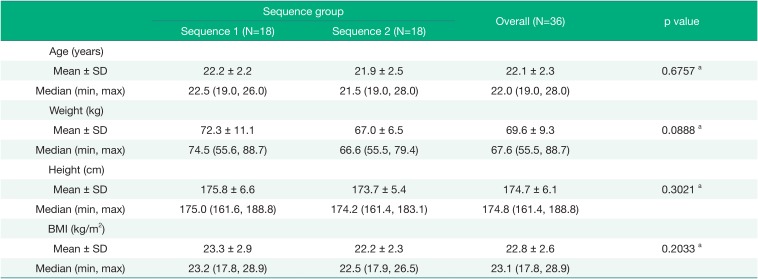
The geometric mean plasma concentration-time curves of solifenacin for the test or reference formulation in healthy male volunteers are shown in Figure 1. The pharmacokinetic parameters of solifenacin in plasma by formulation are summarized in Table 2.
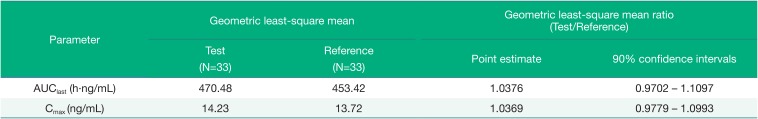
After oral administration of the test or reference formulation, the mean values of AUClast of solifenacin were 486.98 ± 138.47 h·ng/mL and 469.07 ± 128.29 h·ng/mL, respectively. The mean Cmax values of solifenacin were 14.66 ± 3.85 ng/mL and 14.10 ± 3.37 ng/mL, with median(min-max) Tmax values of 5.00(3.00–6.00) hours and 5.00(3.00–6.00) hours, respectively. The mean t1/2 of solifenacin was 33.38 ± 8.06 hours and 32.40 ± 6.29 hours, respectively. The apparent total body clearance and the apparent volume of distribution of solifenacin were very similar for the two formulations.
The point estimates and 90% CIs for the geometric mean ratio (test/reference) of the AUClast and Cmax of solifenacin were 1.0376 (0.9702 – 1.1097) and 1.0369 (0.9779 – 1.0993), respectively (Table 3). The 90% CIs for these pharmacokinetic parameters of solifenacin met the acceptance range of 0.80 – 1.25 for bioequivalence.
Safety
No serious adverse events occurred during the entire study period. A total of 15 AEs occurred in 11 of the 36 subjects after administration of the investigational products. Among these AEs, 14 cases were evaluated as mild, and 1 case was evaluated as a moderate AE in intensity. There were 5 cases of adverse drug reactions (ADRs) in 2 subjects who were treated with the test formulation. The reported ADRs were epistaxis, dry mouth and oropharyngeal pain and all of these ADRs were of mild intensity. There were no significant differences between the test formulation and reference formulation when evaluating laboratory tests, vital signs, physical examinations and ECG results.
Discussion
This study was conducted to compare the pharmacokinetic properties of the test formulation, solifenacin tartrate 10.66 mg, to those of the reference formulation, solifenacin succinate 10 mg. The point estimates and 90% CIs for the geometric leastsquare mean ratio (test/reference) of AUClast and Cmax were shown to meet the bioequivalence criteria.
According to the label for VESIcare® (solifenacin succinate) tablets listed with the FDA, the Tmax of solifenacin is 3 to 8 hours, and t1/2 is approximately 45 to 68 hours after a single dose of 5 to 10 mg tablets.[6] Since solifenacin is a drug with a long half-life and a low CVintra of distribution and clearance, the final sampling time was set to 72 hours according to the guidance instead of using 3 times the t1/2 as the final sampling time.[10111213] In this study, the CVintra values of distribution and clearance were as small as 18.0% and 15.5%, respectively. The t1/2 value in this study was 32.4 hours for the reference formulation and 33.4 hours for the test formulation. The t1/2 value is smaller than that reported in a previous study. This discrepancy could be due to differences in study design; in this study, pharmacokinetic sampling was only performed up to 72 hours post dose. If the final sampling time was set beyond 72 hours, a value similar to that on the label for VESIcare® might have been obtained.
The solifenacin contained in the test and reference formulations was absorbed and reached Cmax at 4.7 hours and 5.0 hours after oral administration, respectively. The plasma concentrations of solifenacin declined over time, with similar slopes for the test and reference formulations. In addition, the 90% CIs for the geometric least-square mean ratios (test/reference) of AUClast and Cmax were within the range of 0.8 to 1.25, thereby satisfying the regulatory criteria for bioequivalence.
In the safety assessment, the AEs were reported 14 cases after the test formulation and 1 case after the reference formulation. However, 10 AEs were deemed to have no causal relationship to the investigational products; only five AEs were decided to be related to the investigational products. All ADRs were of mild intensity and no significant differences was observed in the safety assessments of the formulations.
Our study was conducted in healthy Korean male subjects only who were selected based on inclusion and exclusion criteria. Therefore, further studies in a more general patient population might be useful for extending the applicability of the results of this study.
This study showed that the newly developed tablets containing solifenacin tartrate 10.66 mg are bioequivalent to the existing solifenacin succinate 10 mg and thus meet the established MFDS regulatory criteria. These results suggest that the test formulation is interchangeable with the reference formulation.
 XML Download
XML Download