

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Hepatosplenic T-cell lymphoma (HSTL) is an aggressive extranodal lymphoma that originates from cytotoxic γδ T cells [1, 2]. HSTL patients typically present with hepatosplenomegaly in the absence of lymphadenopathy [1, 2]. Morphologically, HSTL is characterized by sinusoidal infiltration of the spleen, liver, and bone marrow (BM) with monotonous medium-sized neoplastic lymphoid cells; isochromosome 7q is a recurrent cytogenetic abnormality detected in most HSTL cases [1, 2]. The typical immunophenotype of HSTL is as follows: CD2+, sCD3+, CD4–, CD5–, CD7+, CD8–, and T-cell receptor (TCR)γδ+ [1]. However, there have been reports of rare HSTL cases with loss of sCD3 and TCRγδ, posing a major challenge in establishing an accurate diagnosis and determining relapse [3–6]. We report a rare case of HSTL with an unusual immunophenotype characterized by loss of sCD3 and TCRγδ expression at relapse. Unlike previously reported cases, the abnormal cells in our case were positive for CD16 and CD56 and, therefore, could be easily misinterpreted as natural killer (NK) cells. The Institutional Review Board of Samsung Medical Center, Seoul, Korea, approved this study (approval number: 2020-07-154) and waived the need for informed consent.
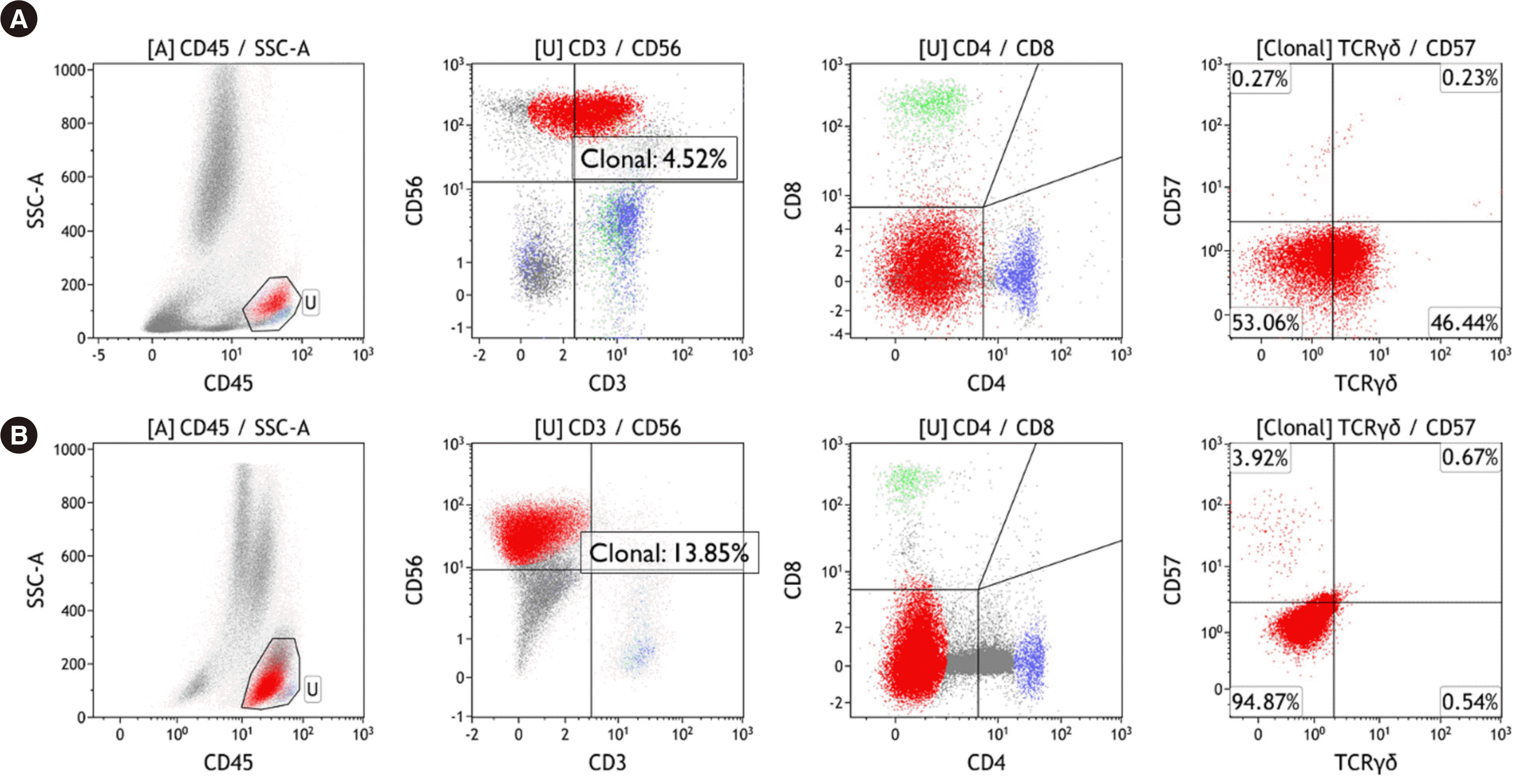
A 57-year-old man visited Samsung Medical Center in September 2019 for fever of unknown origin (FUO). The complete blood count (CBC) result was as follows: hemoglobin, 81 g/L; white blood cell count, 3.38×109/L (without definite abnormal lymphoid cells); and platelet count, 204×109/L. The abdominopelvic computed tomography scan revealed hepatosplenomegaly, with a spleen size of 16 cm. During the FUO workup, a BM biopsy was performed, which led to the diagnosis of mature γδ T-cell lymphoma without an identifiable primary focus. In the initial study, the BM aspirate showed 5–10% of medium-sized abnormal lymphoid cells with pleomorphic nuclei and prominent nucleoli. Flow cytometry was performed using a FACSLyric flow cytometer (Becton Dickinson, San Jose, CA, USA) after staining the BM mononuclear cells with monoclonal antibodies against T- and NK-cell associated antigens, and data were analyzed using the Kaluza software (Beckman Coulter Inc., Miami, FL, USA). Flow cytometry revealed an abnormal T-cell population with the following immunophenotype: sCD3+, cCD3+, CD4-, CD8-, CD56+, CD16+, CD2+, CD5–, CD7+, CD94+, CD57–, and TCRγδ+ (Fig. 1). Chromosomal analysis indicated a normal karyotype (46,XY[20]), and a TCR rearrangement assay revealed clonality in both TCRγ and TCRβ.
The patient underwent multiple cycles of chemotherapy with four different regimens. During the course of chemotherapy, splenectomy and cholecystectomy were performed, and spleen pathology revealed γδ T-cell lymphoma. A follow-up BM study was conducted six months after the initial diagnosis, and the CBC result was as follows: hemoglobin, 105 g/L; white blood cell count, 10.22×109/L (10% abnormal lymphoid cells); and platelet count, 244×109/L. The BM aspirate showed 20% abnormal lymphoid cells, and their immunophenotype, determined by flow cytometry, was sCD3–, cCD3+, CD4–, CD8–, CD56+, CD16+, CD2+, CD5–, CD7+, CD94+, CD57–, and TCRγδ– (Fig. 1); in contrast to the initial findings, loss of sCD3 and TCRγδ was noted. Two possibilities were considered: 1) an unusual immunophenotypic shift to abnormal T cells lacking sCD3 and TCRγδ and 2) an increased proportion of reactive NK cells. A positive TCR rearrangement result supported the former possibility; next-generation sequencing (NGS) was not performed. Furthermore, chromosomal analysis revealed a complex karyotype, including isochromosome 7q (46,XY,add(4)(p16),+6,i(7)(q10),der(10;14)(q10;q10),del(14)(q32)[9]/46,idem,add(X)(p21),-add(4),+4[7]/46,XY[4]), and a specific diagnosis of HSTL was finally made. Although the role of isochromosome 7q in HSTL remains unclear, it has been suggested that HSTL tends to gain an extra isochromosome 7q during the course of the disease, which may promote the proliferation of abnormal cells [7]. The patient expired two months after relapse.
There are only two reports on the loss of sCD3 and TCRγδ in HSTL during follow-up [3, 6]. Detailed information regarding each case is presented in Table 1.
Compared with previous cases, determining disease relapse was very challenging in the present case because of CD56 positivity and simultaneous loss of sCD3 and TCRγδ expression, which made the abnormal lymphoid cells from this case difficult to distinguish from reactive NK cells. In such cases, a meticulous review of cell morphology, cytogenetic studies, and molecular studies should be conducted. Morphologically, immunohistochemistry for granzyme B and perforin could be useful, as these proteins are not expressed in HSTL but are expressed in aggressive NK-cell leukemia (ANKL) [2, 4]. Conversely, HSTL cases could be positive for TIA-1 expression [2, 4]. Identification of isochromosome 7q and TCR rearrangement may also aid the differential diagnosis of HSTL versus ANKL [2, 4]. Due to the limited number of studies, the clinical significance of sCD3 and TCRγδ loss remains to be elucidated. However, one report suggested that sCD3 and TCRγδ loss are associated with poor prognosis [3].
While flow cytometry provides powerful insights into neoplastic cell lineage, it may be a double-edged sword in cases where lineage-specific markers are aberrantly lost, since it could lead to the misclassification of cell lineages. Hematopathologists should always consider the possibility of unusual immunophenotypic changes. Since only a few such cases have been reported to date, the fact that HSTL can present without expression of sCD3 and TCRγδ is not well known. Hence, we present this diagnostically challenging case to share our experience and highlight the fact that T-lineage lymphoma cells with loss of T-cell markers could be mistakenly identified as NK cells in flow cytometry.
 XML Download
XML Download