

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Structural hemoglobinopathies are the most common monogenic disorders globally [1]. Most hemoglobin (Hb) variants are clinically silent but can be identified during neonatal screening, in population studies, or during the quantification of glycated Hb (HbA1c) [2]. The presence of an Hb variant may interfere with HbA1c quantification.
We report a new Hb variant found in a 76-year-old Spanish woman from Murcia. The patient was scheduled to undergo cataract surgery in the left eye in February 2015. As part of preoperative examinations, patient’s blood was collected in three 5 mL EDTA-coated tubes. In cation-exchange (CE)-HPLC in an automated glycohemoglobin analyzer (Bio-Rad D-100; VARIANT, Bio-Rad Laboratories, Hercules, CA, USA), falsely increased HbA1c levels were observed: HbA1c level was detected as 7%, whereas the actual level —measured by affinity chromatography—was 5%. Although no other major peak in CE-HPLC suggestive of a variant was observed, the sample was sent to the hemoglobinopathies referral center at Hematology service, Erythropathology section, Hospital Clínico San Carlos de Madrid, to rule out interference due to a structural hemoglobinopathy. The study did not need approval by the Institutional Review Board, but informed consent was provided by the patient. Experiments were conducted according to the Declaration of Helsinki.
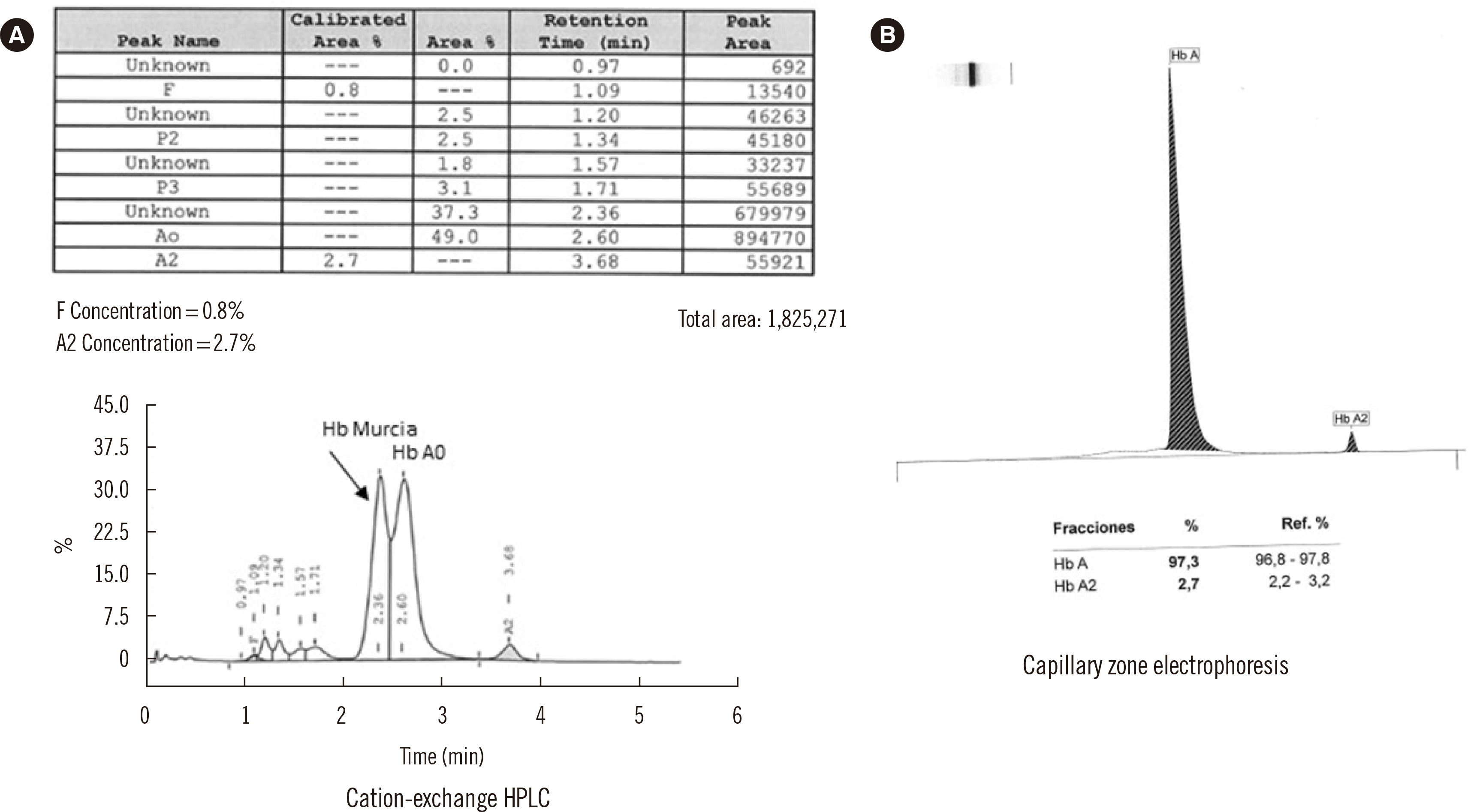
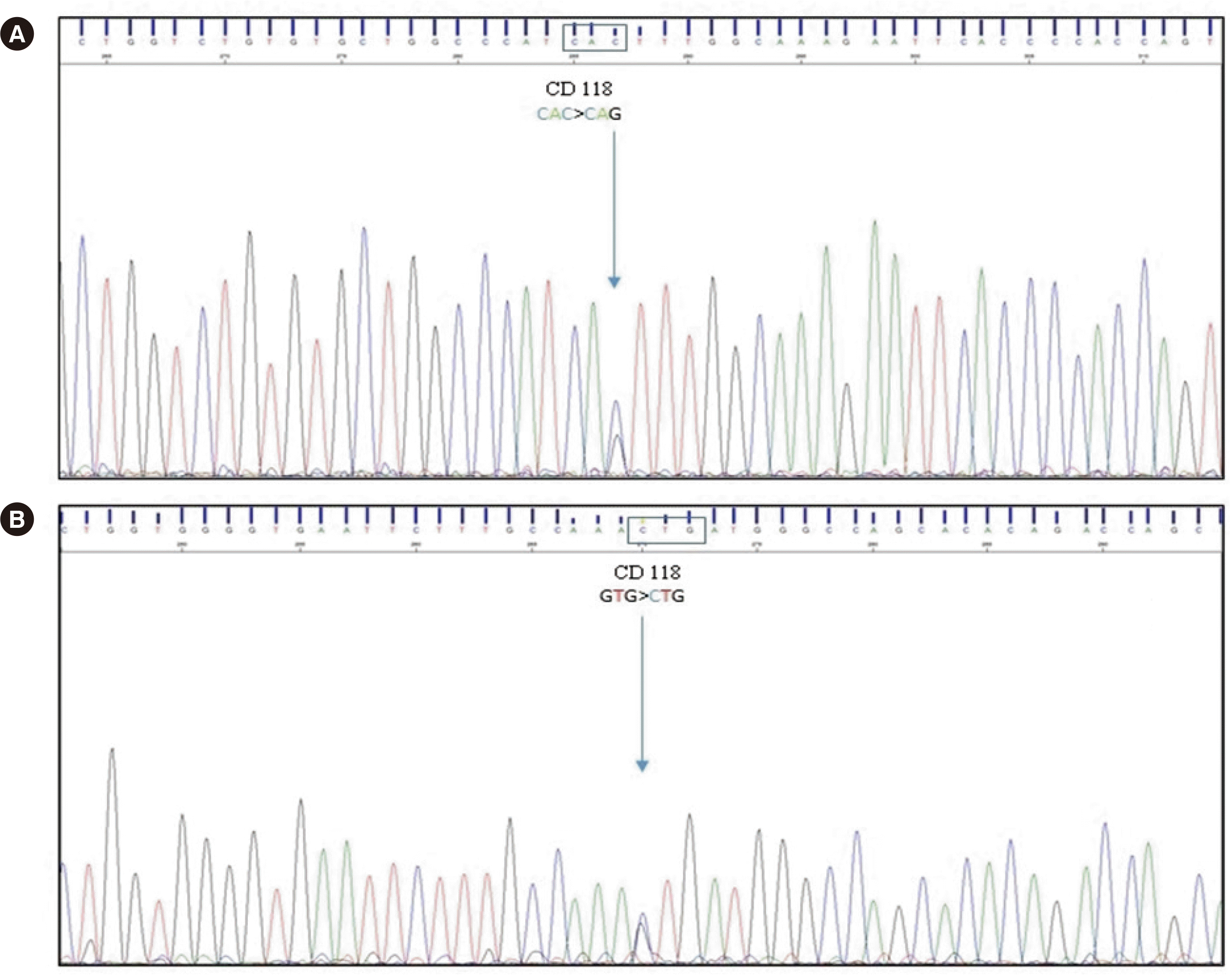
The patient’s hematological findings were normal, with no anemia (Hb 124 g/L [reference range, 120–156 g/L]), no microcytosis (mean corpuscular volume 97.6 fL [reference range, 81–98 fL]), no hypochromia (mean corpuscular Hb 30.7 pg [reference range, 26–33 pg]), and a normal reticulocyte count (0.6%); a peripheral blood smear did not show morphological abnormalities. Hb analysis by capillary zone electrophoresis (Sebia Capillarys Flex Sebia, Norcross, GA, USA) revealed no abnormal peaks (Fig. 1). However, chromatographic analysis by CE-HPLC using the β-thalassemia short program (VARIANT, Bio-Rad Laboratories) revealed the presence of an Hb variant at retention time 2.36 minutes, which accounted for 37.3% of the total Hb. This variant eluted ahead of HbA0, without full separation (Fig. 1). HbA2 and fetal (HbF) levels measured by CE-HPLC were normal (2.7% and 0.7% respectively). The variant was characterized by automatic sequencing of the β globin gene (HBB) as previously described [3]. The sequencing results revealed a transversion of cytosine to guanine (CAC>CAG) in codon 118 of the third exon (Fig. 2), resulting in a missense variant with a glutamine instead of a histidine (HBB:c.354C>G; β118(G19)His>Gln). We named this variant “Hb Murcia” after the city where the patient lives.
Some residues of the C, G, and H helices of Hb are a part of the α1β1 contact area. Most of these contacts are weak, non-covalent bonds, such as Van der Waals interactions and hydrogen bonds, although the latter have a less prominent role in α1β1 contact. These contacts form a rigid dimer, so that during Hb oxygenation and deoxygenation, movement in this area is extremely limited [1]. Therefore, any constitutional variation could give rise to structural modifications that can cause Hb precipitation in erythropoietic progenitors depending on the location of the residue within the helix, its participation in Hb functionality, and the characteristics of the residue. This, in turn, may cause ineffective erythropoiesis or hemolytic anemia. An example of these behaviors is Hb variants at position G19. Position 19 of the G helix of the globin β chain is occupied by residue 118, a histidine. This residue is located externally in the three-dimensional protein structure and interacts with two residues of the α1 globin chain (α110 alanine [G17] and α114 proline [GH2]) and one residue of the same β globin chain (Glu β22 [B4]) through hydrogen bonds [4]. At this position, five variants have been described: Hb P-Galveston, Hb Brent, Hb Tsukumi, Hb North York, and Hb Saitama. All these variants were detected incidentally through electrophoretic and chromatographic methods, as the mobility of Hb is altered due to modification of the surface charge when a positively charged polar amino acid, such as histidine, is substituted with a neutral or negatively charged one. The functional properties and stability of the resulting Hb have been studied for three of these variants. Hb Saitama leads to hemolytic anemia, as it has decreased affinity for oxygen and is unstable—probably because of the insertion of a proline, which tends to have a destabilizing effect on the chain [5]. By contrast, Hb P-Galveston and Hb Tsukumi are functionally normal. The remaining variants, including the one reported here (Hb Murcia), have not been studied for functionality and stability. However, given the hematological profiles of the patients, both the stability and the functionality of hemoglobin are probably not altered and, therefore, these variants are not harmful, i.e., they are clinically silent [3, 6-8].
The redundancy of the genetic code is not the same for all amino acids; while leucine, serine, and arginine are encoded by six different codons, histidine is encoded by only two (CAT and CAC). Accordingly, the probability of finding more variants is greater in the latter. Indeed, Hb Murcia is the sixth variant described at position β118 (G19). All possible variants have been described, except the one that leads to a leucine substitution (CAC>CTC). Genetic coordinates of HBB gene are given in the hg19 genome version. The nucleotide cytosine HBB:c.354C>G variant (transcript HBB [NM_000518.5C>G]) is highly conserved among primates and placental mammals, as evidenced by the value of +2.035 assigned in the 100 vertebrates Basewise Conservation by PhyloP (phyloP100wayAll), where positive values indicate evolutionary conservation of a nucleotide [9]. The variant chr11:5225688C>G (β118(G19)His>Gln) is classified as pathogenic by UniProt Variants, and this classification has been confirmed according to the American College of Medical Genetics and Genomics guidelines [10].
Hb Murcia is a new clinically silent Hb variant. Knowledge of the existence of this variant is important because it could interfere with the results of glucose monitoring by CE-HPLC, wherein glycosylated Hb Murcia co-elutes with HbA0, thus preventing the correct estimation of HbA1c. When the abnormal glycosylated fraction is not separated, HbA1c would be overestimated; therefore, the HbA1c chromatogram should be carefully reviewed to detect the possible presence of variants that interfere with the measurement. In such a case, clinicians would have to use other methods for correct quantification and to avoid potentially inappropriate treatments.
 XML Download
XML Download