

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Human bone is a highly active organ that maintains its homeostasis through an exquisite balance between bone formation by osteoblasts and bone resorption by osteoclasts. The energetic balance between these two cell types results in bone remodeling [1234]. Imbalance of free radical production and redox mechanisms can result in oxidative stress, which damages cellular components and their functions. Oxidative stress is responsible for the aging process and many diseases such as bone fracture, bone diseases, and osteoporosis [5]. The role of free radicals and oxidative stress in bone remodeling is unknown [6].
There is a clear difference between reactive oxygen species (ROS) required for basic cellular mechanisms and excessive ROS production that might cause oxidative stress and contribute to the pathogenesis of major diseases such as diabetes, neurodegeneration, and cancer [7]. Among the various ROS, hydrogen peroxide (H2O2) is perhaps the most ubiquitous of these species, which is found at measurable levels in all animal tissues. H2O2 is very stable and can reach molecular targets distant from its site of generation. Because H2O2 is a small, uncharged molecule, it easily crosses cell membranes and localizes in multiple subcellular compartments [8]. The effects of H2O2 are concentration-dependent and range from physiological signaling in cell proliferation, migration, survival, differentiation, and gene expression [9101112] to overt cell death [131415].
Propofol (2,6-diisopropylphenol) is an intravenous sedative-hypnotic agent used for general anesthesia and sedation of patients in intensive care unit [1617]. Propofol has a similar structure to the endogenous antioxidant vitamin E and exhibits antioxidant activities [1819]. In other studies, administration of anesthetic agents was found to result in pharmacologic preconditioning against oxidative injury [2021].
Autophagy is a conserved cellular catabolic process that can engulf cytoplasmic components and degrade them through the lysosomal pathway, thus, they can then be recycled. Generally, autophagy is thought to be induced under stress conditions [222324]. However, whether the induction of autophagy contributes to cell survival or cell death remains elusive. Oxidative injury is one of the most common causes of bone remodeling inhibition. Therefore, a sustainable drug is needed to identify better, safer anabolic agents with low cytotoxicity that act by either increasing osteoblast proliferation or differentiation to enhance bone formation [25]. Several lines of evidence have shown that anesthesia drugs may have a beneficial effect on bone loss and fracture outcomes [26]. On the basis of this, it was hypothesized that the antioxidant properties of propofol may protect human fetal osteoblast (hFOB) cells against oxidative injury caused by H2O2. Therefore, the aim of this study was to evaluate the protective effects of propofolmediated autophagy activation on oxidative stress induced by H2O2 treatment in the hFOB cell line.
MATERIALS AND METHODS
1. Reagents
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma (St. Louis, MO, USA). 3-Methyladenine (3-MA, a class III PI3K inhibitor) was obtained from Calbiochem (La Jolla, CA, USA). Antibodies against collagen type I, bone morphogenic protein (BMP)-2, osterix, and transforming growth factor-beta (TGF-β1) were purchased from Abcam (Cambridge, UK). The GAPDH, mouse antirabbit IgG, and rabbit anti-mouse IgG antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals and reagents were purchased from Sigma unless otherwise specified.
2. Cell culture
Human fetal osteoblast cell line (hFOB 1.19) was purchased from ATCC (Rockville, MD, USA). Cells were cultured in Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12) containing 4 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, and 1.0 mM sodium pyruvate supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (GIBCOBRL, Rockville, MD, USA). Cells were incubated at 37℃ in a humidified 5% CO2 –95% air incubator.
3. Propofol treatment
The propofol stock (56 mM) was kept at room temperature until use and diluted to the proper concentration with DMEM/F-12 when needed. Prior to propofol treatment, cells were grown to about 75% confluence and then exposed to propofol at various concentrations (0, 3, 30, 100, or 300 µM) for 2 h. The cells were divided into the following groups: control cells were incubated at 37℃ in a humidified atmosphere with 5% CO2 and without any additional treatment. H2O2 cells were exposed to 200 µM H2O2 for 2 h. PPC/H2O2 cells were pretreated with 100 µM propofol for 2 h and then exposed to 200 µM H2O2 for 2 h. 3-MA/PPC/H2O2 cells were pretreated with 1 mM 3-MA and 100 µM propofol for 1 h and 2 h, respectively. After treatment of 3-MA and propofol, cells were exposed to 200 µM H2O2 for 2 h. The experimental paradigm as just described is visualized in Fig. 1.
4. Cell viability assay
The cell viability of hFOB cells was determined using an MTT assay. Cells were cultured in 96-well plates (4 × 103 cells/well). The cells were then treated with the indicated concentrations of drug. At the end of the treatment, 100 µL of MTT solution (500 mg/mL) was added to each well. The cells were incubated for 4 h at 37℃. The formazan crystals were then solubilized in DMSO (200 µL/well) by constant shaking for 15 min. The cell viability was measured by an ELISA reader (Tecan, Männedorf, Switzerland) by determining the wells' absorbance at 620 nm.
5. Fluorescence microscopy
Cells were harvested and cytocentrifuged onto a clean glass slide. Cells were stained in 1 µg/mL Hoechst 33342 for 15 min at 37℃ in the dark and subsequently washed in phosphate buffered saline (PBS). The slides were mounted with glycerol. The samples were observed and photographed under an epifluorescence microscope (Carl Zeiss, Goettingen, Germany). Cells were grown on coverslips and treated with propofol. After 24 h, cells were stained with 0.05 mM monodansylcadaverine (MDC), a selective fluorescent marker for autophagic vacuoles, at 37℃ for 1 h. The cellular fluorescence changes were observed using a fluorescence microscope (Axioskop, Carl Zeiss, Germany). For further detection of the acidic cellular compartment, we used acridine orange (AO), which emits bright red fluorescence in acidic vesicles but fluorescence green in the cytoplasm and nucleus. Cells were stained with 1 µg/ml AO for 15 min and washed with PBS. Acidic vesicular organelles (AVOs) formation was obtained under a confocal microscope LSM 700 (Carl Zeiss, Germany).
6. Flow cytometer analysis
Quantification of apoptotic cells was determined by Annexin V-FITC/PI staining. Adherent cells were collected by trypsinization and centrifugation, and then were resuspended in 500 µL 1X binding buffer. Then they were stained with 5 µL Annexin V-FITC and propidium iodide (PI) (50 µg/mL) and were incubated at room temperature for 5 min in the dark. The cells were analyzed with a CYTOMICS FC500 flow cytometer system (Beckman Coulter, CA, USA). The results were shown as quadrant dot plots with intact cells (Annexin V-/PI-), early apoptotic cells (Annexin V+/PI-), late apoptotic cells (Annexin V+/PI+), and necrotic cells (Annexin V-/PI+).
7. Alizarin red S staining
Mineralization of hFOB cells was determined in 24-well plates using Alizarin Red S (Sigma, St. Louis, MO, USA) staining. The cells were washed once with distilled water (DW) after fixation with 4% paraformaldehyde (PFA) and were stained with 2% Alizarin Red solution according to the manufacturer's protocol. For quantification, absorbance of the released Alizarin Red was measured at 550 nm with a microplate reader.
8. Western blot analysis
Cells (1.5×106) were washed twice in ice-cold PBS, resuspended in 200 µL ice-cold solubilizing buffer (300 mM NaCl, 50 mM Tris-HCl (pH 7.6), 0.5% Triton X-100, 2 mM PMSF, 2 µL/mL aprotinin, and 2 µL/mL leupeptin), and incubated at 4℃ for 1 h. The lysates were centrifuged at 13,200 rpm for 30 min at 4℃. Protein concentrations of cell lysates were determined using a Bradford protein assay (Bio-Rad, Richmond, CA, USA), and 20 µg protein were loaded per lane and resolved by 10% SDS/PAGE. Gels were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking, the membranes were reacted with the appropriate primary antibodies. Immunostaining with secondary antibodies was detected using Super-Signal West Femto substrate (Pierce, Rockford, IL, USA).
RESULTS
1. Effect of propofol treatment on cell proliferation
The effect of propofol on hFOB cells was investigated over a wide concentration range. We pretreated hFOB cells with various doses of propofol, exposed the cells to oxidative stress, and then measured cell viability by an MTT assay. The viability of propofol-treated hFOB cells increased in a dose-dependent manner. Propofol and 3-MA alone did not show any significant toxic effects on hFOB cells (Fig. 2A, 2B). Incubation with 200 µM H2O2 markedly decreased cell viability, which was significantly attenuated when treated with propofol, suggesting that propofol abated H2O2-induced oxidative stress in osteoblasts (Fig. 3).
2. Propofol protected against H2O2-induced apoptosis in hFOB cells
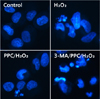
The effect of propofol on apoptosis was examined by Hoechst 33342 staining of hFOB cells exposed to control, H2O2, PPC/H2O2, and 3-MA/PPC/H2O2. Cells were viewed under a fluorescence microscope (400X magnification) (Fig. 4). The majority of hFOB cells in the control and PPC/H2O2 groups showed normal morphology with round regular nuclei. In contrast, apoptotic bodies were seen in the H2O2 group and the 3-MA/PPC/H2O2 group cells. Pretreatment with propofol effectively reduced hFOB cell apoptosis according to the restored morphology. Annexin-V FITC/PI staining quantitatively confirmed the antiapoptotic effects of propofol (Fig. 5). Compared with control group, the portion of Annexin-V(+)/PI(−) cells in H2O2 group increased from 3.4% to 10.3% (P < 0.05). However, pretreatment with propofol 2 h prior to PPC/H2O2 significantly attenuated the percentage of Annexin-V(+)/PI(−) cells to 1.2% (P < 0.05), demonstrating the anti-apoptotic effect of propofol.
3. Effect of propofol treatment on autophagy activation
Prominent accumulation of autophagy-specific MDC staining was observed around the nuclei in the PPC/H2O2 group (Fig. 6). Similarly, after AO staining, red fluorescent spots appeared in PPC/H2O2 group hFOB cells, while the control and 3-MA groups showed mainly green cytoplasmic fluorescence (Fig. 7).
4. Effect of propofol preconditioning on bone nodular mineralization
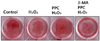
Abundant bone nodular mineralization occurred in the control group, and propofol preconditioning had no influence on this. Cells in the PPC/H2O2 group showed higher bone nodular mineralization compared to the control group and bone nodular mineralization increased gradually with increasing propofol preconditioning (Fig. 8).
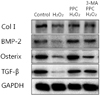
5. Effect of Propofol preconditioning on collagen type I, BMP-2, osterix, and TGF-β 1
Propofol preconditioning was found to increase the expression of bone-related proteins such as collagen type I, BMP-2, osterix, and TGF-β1 (Fig. 9). These results suggest that pretreatment with propofol induces the expression of collagen type I, BMP-2, osterix, and TGF-β 1 in osteoblasts under H2O2 injury.
DISCUSSION
We demonstrated that propofol preconditioning increased the proliferation, differentiation, and maturation of osteoblasts during oxidative injury in our study. The healing process is very important to recover from fractures, and delayed healing after bone implants can cause premature loosening or failure. In our cell viability analysis, there is a distinction between our study and a previous study, which reported an inhibitory effect of non-steroidal anti-inflammatory drugs on fracture healing [27].
In this study, we demonstrated that propofol preconditioning has positive effects on the expression of collagen type I, BMP-2, osterix, and TGF-β1. BMP-2 and TGF-β1 play a crucial role in inducing osteoblast differentiation and bone formation at the preosteoblast stage [2829]. These proteins play an important role in the differentiation of osteoblast progenitor cells, with significant upregulation observed in both matrix synthesis and mineralization [303132]. Collagen type I plays a crucial role in cell adhesion, proliferation, and differentiation of osteoblasts [33]. Osterix is a transcription factor that regulates important osteoblast genes such as collagen type I, osteocalcin, and osteopontin [34].
The results of our study reveal a new direction of research on the mechanisms of propofol-mediated cytoprotection in oxidative injury. Because of the antioxidative effect of propofol, propofol preconditioning enhanced autophagic activity in hFOB cells under oxidative injury. Mechanisms underlying these effects of propofol may be largely attributed to the deactivation of mitochondrial stress pathways.
Chen et al. [35] suggested that ROS play a key role in the regulation of autophagy. In this study, propofol preconditioning not only decreased H2O2-induced oxidative stress, but also activated autophagy. Many studies have focused on oxidative injury in cells, but agents that could effectively protect against oxidative injury remain unidentified. The relationship between apoptosis and autophagy is complex. Autophagy represents a stress adaptation for inhibition of cell death, however in other cellular settings, autophagy represents an alternative pathway to autophagic cell death [3637].
This study shows that propofol pretreatment increases the osteoblast proliferation rate. No functional studies were performed to investigate the effects of propofol on the bone healing process. Therefore, although the findings of this study are limited to an in vitro interpretation, we suggest that propofol may have a beneficial effect in the recovery from bone stress injury.






 XML Download
XML Download