

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Next-generation sequencing (NGS) has facilitated rapid growth in the development of targeted therapies once it was adopted by research institutions and clinical laboratories to elucidate the mutational profiles of cancers [12]. Several clinical trials targeting specific variants have been performed worldwide, and many new candidate genes have been suggested as specific markers for particular diseases through NGS-based tests [13]. Researches using NGS on the number of somatic and epigenetic variants have increased understanding of the pathophysiology of cancers [1456]. Moreover, confirmation of gene variants is also a major criterion for the diagnosis of hematologic malignancies [7]. The importance of the accurate detection of specific gene variants has been emphasized for precision medicine in a number of cancers including hematologic malignancies [2]. The need to identify mutational profiles for use in precision medicine was sufficient for the incorporation of NGS tests into clinical laboratories; however, the setup and validation of these tests for variant detection in malignancies is challenging in clinical laboratories for several reasons. First, owing to tumor heterogeneity and mutational complexity, robust validation is needed to guarantee test accuracy, especially for low variant allele frequency (VAF) variations [8]. Second, a large amount of data needs to be carefully analyzed and interpreted to meet the QC metrics thresholds. Because of the complex workflow of NGS testing, in both wet and dry laboratories, experts in clinical genetics and bioinformatics as well as technicians proficient in molecular genetic testing are necessary. We suggest practical guidelines for validating NGS-based somatic panels for the diagnosis of hematologic malignancies.
GENERAL CONSIDERATIONS FOR TEST DEVELOPMENT
The clinical purpose of the NGS-based somatic panels must first be determined. This could include molecular diagnosis, detecting therapeutic targets, or monitoring minimal residual disease (MRD). Recently, many driver mutations have been identified in mutational profiles and used to develop NGS gene panels for hematologic malignancies [34568911]. Currently, targeted NGS-based somatic panels for hematologic malignancies may be laboratory-developed or commercially available. The genes in the designed panel should be selected after considering the clinical relevance and characteristics of the target genes. Careful consideration is required in the initial gene selection as re-validation would be necessary even if a small subset of genes is changed or added.
Platform selection
The platform used to perform NGS tests should be chosen considering all aspects, including cost, user accessibility, turnaround time, test performance, data quality, expected errors, available bioinformatics tools, and commercial gene panels of interest. Several platforms have been developed for clinical diagnosis. Currently, the two main platforms used in Korean clinical laboratories are MiSeq or NextSeq (Illumina, San Diego, CA, USA) and Ion Torrent (Thermo Fisher Scientific, Waltham, MA, USA). Illumina platforms use reversible terminator-based sequencing with optical detection of fluorescently labeled nucleotides [12]. Ion Torrent platforms use non-optical semiconductor sequencing with unmodified nucleotides [12]. These platforms use hybrid capture or amplicon-based methods in the target enrichment process. Capture-based methods usually provide even coverage of target sequences with good reproducibility but require higher amounts of DNA and a longer run time; they are usually errorprone in GC-rich regions [121314]. In contrast, the advantages of amplicon-based methods are that they require a shorter run time and lower amounts of DNA; however, primer dimers or non-specific amplification products can be generated [121314]. Once the characteristics of each platform are understood, platforms that suit the actual conditions of a particular clinical laboratory should be selected.
Designing or choosing a gene panel
The panel and targeted genes should be determined based on clinical purpose and disease category. The extent of a disease category among hematologic malignancies needs to be determined; genes related to only myeloid neoplasms or all categories of hematologic malignancies are included in the panel. Targeted regions should be determined based on the locations and characteristics of clinically significant variants for diagnosis and therapeutic decisions. According to the WHO classification, JAK2, CALR, MPL, and CSF3R are crucial genes for the diagnosis of myeloproliferative neoplasms [15]. In addition to balanced translocations/inversions, gene variants, such as NPM1, CEBPA, RUNX1, FLT3, IDH1/2, ASXL1, and KIT, are also important diagnostic and prognostic markers in acute myeloid leukemia [1115]. Moreover, MYD88 and BRAF should be included in the diagnosis of a specific type of lymphoma [11]. The reportable range should also be determined considering the specific characteristics of the sequence and type of variants. The areas of targeted regions below the minimum coverage need to be excluded from the reportable range and documented if the excluded regions are clinically important [161718]. When using a commercial panel, it is necessary to verify the anticipated test performance of the target regions in each laboratory.
VALIDATION OF A GENE PANEL FOR HEMATOLOGIC MALIGNANCIES
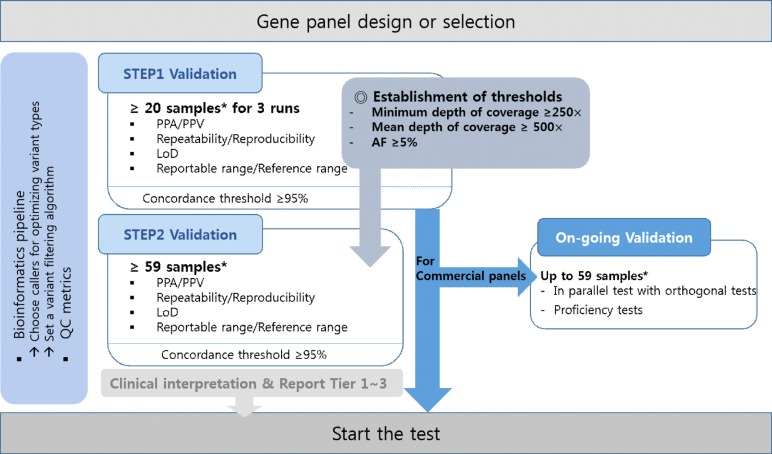
A two-step approach is recommended for validation (Fig. 1). The first step of validation (Step 1), known as pilot tests, is necessary for optimization and checking for possible errors during the entire NGS testing process. After confirming >95% concordance rate with known variants and meeting the QC metrics thresholds, the second step of validation (Step 2) is conducted with established thresholds of essential parameters, such as depth of coverage and VAF, for each type of variant. When using commercial panels, laboratories can perform ongoing validation instead of Step 2. Ongoing validation is described in a separate section.
General considerations for validation
Samples
Sample types should be determined prior to validation; each sample type to be used in practice should be included in the validation process. Whole blood (WB) or bone marrow (BM) samples are the most commonly used for hematologic malignancies. However, other samples, including formalin-fixed paraffin embedded (FFPE) tissues, various body fluids, cell-free DNA, and skin tissue (skin fibroblasts for germline DNA), can also be used [19]. To examine a particular type of sample, the validation step should be performed for multiple samples of the same type. Adequate purity, volume, and proper storage conditions (e.g., time and temperature) of each sample are necessary for optimal testing. Approximate VAFs of known variants (from previous Sanger testing or NGS testing) in each sample would be useful for validation design. Fresh WB and BM samples are usually considered best for practical NGS testing because of the relatively high quality and quantity of DNA and RNA from neoplastic cells. We discuss the type of samples, mainly focusing on WB and BM. In several cases, reference materials (RMs) or commercial controls can also be included during validation. Pooling of up to three samples with different variants is also viable and can be regarded as three samples. If the samples originated from different patients, those with the same variant can be used in <10% of the total samples for validation.
Type of variants
Frequent variants with clinical significance should be included as positive samples during validation. In a panel of myeloid malignancies, c.1849G>T (p.V617F) in the JAK2 gene or an internal tandem duplication in the FLT3 gene are included in the validation with high priority [20]. The variant type should be determined before implementing validation. We mainly describe single-nucleotide variants (SNVs) and insertions and/or deletions (indels), as the accurate detection of the other types of variants is challenging in clinical diagnosis.
Number of genes in the panel required for validation
There is no consensus regarding how many genes should be included in the panel undergoing validation. Validating many regions would be good for reliability; however, it is nearly impossible in many clinical laboratories. Clinically relevant variants should be included as a priority, as previously described in the section of designing or choosing a gene panel. Samples with two or more known variants, commercial controls, or cancer cell lines could be efficiently used to reduce the number of samples required for validation [1617].
Step 1 validation: pilot test of a custom panel or verification of a commercial panel
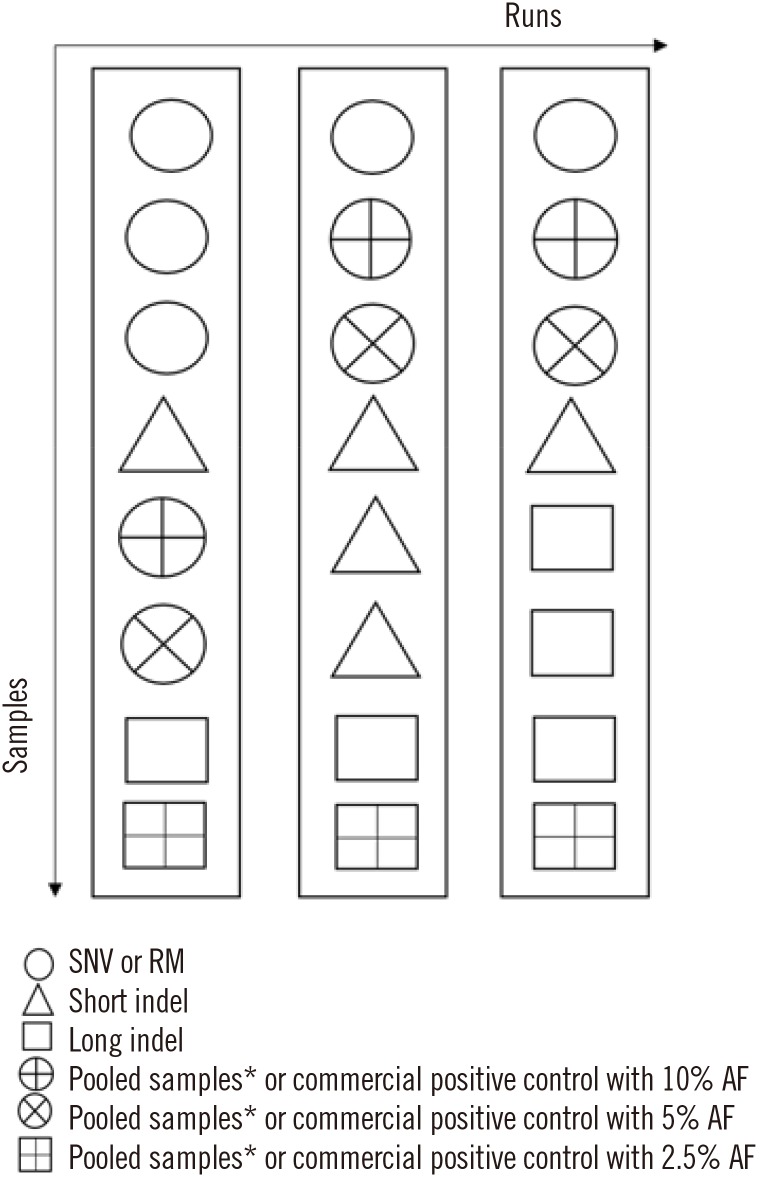
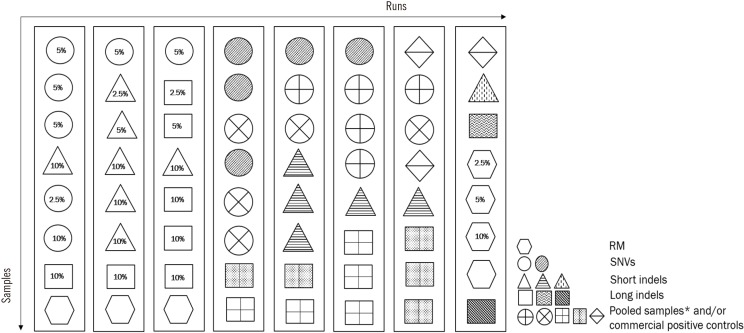
Step 1 (pilot tests or verification) can provide an overview of initial validation before Step 2. To optimize a test or to verify the performance of a commercial panel, the entire process should be evaluated by Step 1 validation [131621]. Unexpected problems are often identified and corrected during this step. We recommend that at least 20 samples and at least three runs be included and performed to evaluate the NGS testing performance parameters, such as accuracy, precision, and limit of detection (LoD), for each type of sample and variant (Fig. 2). Mixing a number of samples with known variant burden is a means of validating multiple variations with fewer samples. Desired VAF thresholds, depth of coverage for each position, and mean depth of coverage should be established in Step 1 validation at given QC metrics thresholds [39]. We recommend that 5% VAF, a minimum coverage of 250 reads for each position, and a mean coverage of 500 reads should be the threshold for a gene panel for hematologic malignancies [1316]. These analytical goals applicable to Steps 1 and 2 (including ongoing validation) require validation with minimum 20 and 59 samples, respectively. Different VAF thresholds can be adopted for different variants. Uncovering common variants, such as an indel in the CEBPA gene, may be challenging with a low depth of coverage and low VAF in accordance with highly GC-rich regions. In this case, systematic errors should be discriminated and documented, which may be reviewed when designing a panel [1416]. A concordance rate >95% should be met for the detection of known variants through NGS and Sanger sequencing before Step 2 or before beginning to test patients using the panels.
NGS performance characteristics
Positive percentage agreement (PPA) and positive predictive value (PPV)
The accuracy parameters, such as PPA and PPV, for each type of variant should be established during validation [1]. PPA is the ability of NGS tests to obtain positive results measured in concordance with positive results obtained by an orthogonal test (e.g., Sanger sequencing). PPV is the proportion of the number of positive NGS test results that have the target condition, as determined by the orthogonal test. RMs could constitute a good source of PPA/PPV, providing true positive variants [1316]. In addition to RM-based PPA/PPV, PPA/PPV can be derived from the results of known true variants, including hotspots and non-hotspots, from patient samples. The formulas for calculating PPA and PPV are as follows:
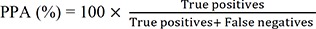
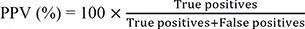
Precision (repeatability and reproducibility)
Repeatability (within-run precision) and reproducibility (between-run precision) should be evaluated. We recommend that at least three samples be used for each type of variant and triplicates for both within-run and between-run precisions should be arranged during validation design [162122]. Using RMs or commercial positive controls for each type of variant may serve as an alternative option.
LoD
The LoD should be evaluated for each type of variant through dilution studies of pure patient samples with an RM at variable percentages (25%, 10%, 5%, 2.5%, or 1%) based on which the minimum VAF can be determined according to the purpose of the panel [17]. We recommend a 5% VAF for each type of variant as adequate for the diagnosis of hematologic malignancies; however, 1% VAF should be used for the MRD panel [23]. As it might be difficult to obtain 5% VAF in certain circumstances, such as long indels, GC-rich regions, and repetitive regions, if these occur in a clinically significant region, they should be documented in the clinical report. When using patient samples, at least three samples for each type of variant should be tested in three independent experiments [24]. Serial dilution of pure patient samples with RM, pooled patients' samples, cancer cell lines, and/or commercial controls comprising all types of variants at a specific VAF can be used for LoD determination [131721].
Reportable range and reference range
The reportable range can be determined based on two factors. First, the range should meet the QC metrics thresholds. Second, the range is determined considering target regions with clinical significance. Limitations should be described for specific regions showing lower than minimal depth of coverage. The reference range can be described as the range of normal sequence variation occurring in the general population [17]. The reference range should be included in the report detailing what types of variants were reported. Recent guidelines for somatic variant interpretation and reporting classify the variants into four tiers [20]. Based on these guidelines, we recommend that tier 1–3 variants should be reported in hematologic malignancies. Additionally, the criteria for confirming detected variants using orthogonal tests should be included in the report. Although providing relatively low to medium sensitivity compared with NGS, confirmatory tests, such as Sanger sequencing, pyrosequencing, quantitative PCR, and multiplex ligation-dependent probe amplification, according to variant type, can be described in the report for clinicians.
Ongoing validation
Ongoing validation would be applicable for commercial NGS-based somatic panels that meet the QC metrics in Step 1. Positive samples based on a panel testing service could be used for other orthogonal methods. We do not recommend parallel testing using samples with negative results. It is difficult to confirm true negative results by Sanger sequencing, which has lower analytical sensitivity. Although a previous study has reported that orthogonal validation is not required [25], confirmation between NGS and Sanger sequencing or other orthogonal methods regarding positive results in a specific gene region should be performed using >59 samples, which represent an adequate proportion of each type of variant. For practical reasons, pooled samples with different variants from different patients are also available (see the sample section in general considerations for validation). Additionally, laboratories using commercial panels should participate in proficiency tests (PT) with reference laboratories. Reference laboratories would qualify by undergoing external quality assessment such as domestic and international PT programs for the relevant disease panels. Reference laboratories should be able to provide positive samples, which would help reduce the burden of collecting positive samples and the cost of validation and/or quality controls, especially at relatively small institutions.
Step 2 validation
Basic parameters, including PPA/PPV, precision (repeatability and reproducibility), LoD, and reference range/reportable range should be validated again (see the performance characteristics section in Step 1) with the desired VAF thresholds, depth of coverage for each position, and mean depth of coverage established in Step 1 at given QC metrics thresholds. Although these parameters are preliminarily validated in Step 1, this step would strengthen the validation with additional samples. Based on previous recommendations of validation for somatic variants, in the next step of validation, a minimum of 59 samples is suggested as an adequate number of samples (after statistical calculation), as it represents <5% analytical sensitivity with 95% confidence and ≤1.9% false-positive rate [16]. Generally, validation needs to be performed using as many samples as known variants among patients. However, it is difficult to collect samples that have clinically relevant variants for certain gene panels. For practical reasons, pooled samples with different variants from different patients are also available (see the sample section under general considerations for validation). An example for the design of Step 2 validation is shown in Fig. 3.
Considerations for bioinformatics pipelines
Bioinformatics pipelines or data analysis pipelines include the following steps: read alignment, variant calling, variant annotation and reporting, and generation of QC matrices. The general rules are as follows: for test performance, different computational approaches and validation processes are needed for different classes of sequence variants, namely SNVs, indels, copy number variations (CNVs), and structural variations (SVs). Additionally, software tools should be selected considering the design, purpose, and characteristics of the test. A review of various software or pipelines is beyond the scope of these guidelines. General considerations for selecting software and validation are discussed in this section.
SNVs
For SNV detection, it is necessary to select software specific for somatic SNVs. The software algorithms for constitutional genome analysis can miss variants with VAFs falling outside the expected range for heterozygous and homozygous variants [26]. In cases where low VAF is expected, software optimized for somatic variants is recommended, as it would be optimized for cancer samples with varying levels of tumor purity and heterogeneity [27].
Indels
Indel variants have various size and sequence complexities; thus, accurate alignment, calling, and annotation are technically challenging. During the alignment step, algorithms, including local realignment, should be considered to minimize base pair mismatches. Indels <20 bp can be accurately called by algorithms using probabilistic modeling [16]. However, for accurate detection of medium to large indels (e.g., FLT3-ITD insertion), additional indel-calling algorithms, such as split-read analysis, are required. Various SV callers using split-read analysis are available for detecting long indels; however, most are not fully validated for high-coverage NGS-based somatic panels. Thus, validation of long indel detection with clinical samples is an additional requirement. Furthermore, software characteristics must be considered before installation; for example, some SV callers, such as DELLY and LUMPY, are ideal for detecting duplications or insertions, while Pindel and LUMPY seem to be inferior to SvABA for detecting deletions <300 bp [28].
CNV
Somatic CNV detection in cancer samples is challenging. This limitation makes CNV testing an optional or supportive method. The calling algorithm for CNV is different from those for SNVs or indels in that it is generally inferred based on the read depth data. The most challenging issues for accurate CNV calling are as follows: first, tumor purity and heterogeneity must be taken into account to solve the dilution of CNV signals [27]. However, it is difficult to calculate the purity values, especially in panel sequencing data. Second, batch effect and read-depth normalization should be handled carefully. In contrast, if the CNV calling algorithm is optimized for WB, BM, or fresh tissue samples, it should generally not be applied with additional optimization algorithms for FFPE samples. DNA from FFPE could have different characteristics because of DNA degradation [29], which could cause biased CNV results (both false positive or negative signals).
SVs
There are several limitations for calling SVs. The SV breakpoints are mostly located in non-coding DNA regions, introns, or highly repetitive regions [27]. Therefore, the target regions tend to be too broad and significantly less uniform. These limitations are related to reduced test efficiency and accuracy. In addition, although several tools for SV calling have been introduced, many have yet to be validated or optimized for high-coverage panel sequencing. Validation and parameter optimization are required for each panel; without rigorous validation, including LoD, SV analysis should be used as supportive or optional information.
RNA SVs
RNA SV analyses using NGS have a different test scope from conventional RNA tests; novel fusions or extremely rare fusions that are important for diagnosis and treatment can be detected and analyzed [30]. Many tools for chimeric transcript detection have been introduced. However, each tool has different performance and algorithm [3132]. After reviewing the characteristics of each software, the performance should be validated with true positive and negative samples. To improve the sensitivity and specificity, several software are used for calling, and the results can be integrated and analyzed considering the sensitivity and specificity of the software [3334].
QC metrics
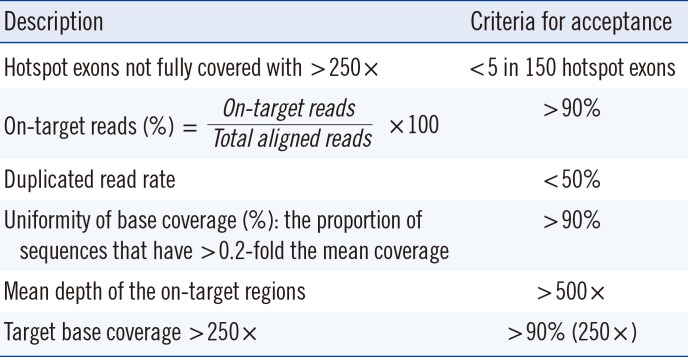
Bioinformatics QC metrics include base and mapping quality scores, on-target reads, duplicated read rate, uniformity of base coverage, mean depth of the on-target regions, target base coverage <250×, numbers and types of variants from the reference, and the transition:transversion ratio in the exome and the genome. These parameters may be modified based on the platform and bioinformatics algorithms; other metrics can also be added. Acceptance criteria are needed for the metrics; most importantly, it is needed to evaluate whether essential target regions, such as mutational hotspots, are fully covered with >250× coverage (Table 1).
DISCLAIMERS
Disclaimers after validation can be divided into three categories: type of sample, specific regions in the panel with technical issues, and interpretations. First, the type(s) of sample(s) used for validation should be described in the validation and the test report document. Additionally, poor conditions related to DNA quality issues should be reported when the tests cannot be performed or when the test results do not meet the established QC parameters. Moreover, high GC content regions, pseudogenes, and repetitive sequences should be documented in the report [131622]. Lastly, analytical sensitivity, such as LoD and reportable ranges, should be specified in the report. The policy for reporting incidental findings of potential pathogenic germline variants should also be described in the disclaimers [135].
CONCLUSIONS
Recently, NGS has been widely used in clinical laboratories; targeted gene panels for hematologic malignancies are being adopted for various purposes. Workflow complexity and the extensive cost and number of samples are main hurdles in the application of somatic panels in the clinical setting. The final goal of NGS testing is reporting reliable results over a minimum quality threshold without Sanger sequencing validation. With the rapid development of technology and bioinformatics tools, we expect this demanding workload of NGS testing validation to be simplified in the near future. The issues related to incidental findings and types of variants other than SNVs and indels need to be discussed in future guidelines. The present guidelines provide general considerations in setting up and validating clinical NGS-based somatic panels in hematologic malignancies. Thus, detailed issues regarding target enrichment method, sequencing platform, and bioinformatics software are beyond our scope and need to be reviewed in a separate paper.

 XML Download
XML Download