

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The routine use of prenatal sonography has made early identification and serial evaluation of congenital lung lesions possible [1]. The antenatal diagnosis of lung abnormalities characterized by cyst formation or increased echogenicity include congenital cystic adenomatoid malformation (CCAM), pulmonary sequestration (PS), bronchogenic cysts and congenital lobar emphysema. These fetuses have a congenital echogenic mass, including a mediastinal shift and displacement of the heart, resulting in compression of the contralateral lung [2]. In some cases, there is acute respiratory distress after birth, requiring the standard treatment of surgical removal of the affected area [3]. In order to improve prognosis in perinatal care, it differentiated diagnosis of lung abnormalities is necessary. Congenital lobar emphysema (CLE) is a rare developmental anomaly of the lung, characterized by air trapping and overdistension of segments and lobes [4,5]. Prenatal diagnosis of CLE has been rarely reported in the literature and only histology later can confirm the presence of CLE [4,6].
Case Report
A 33-year-old, gravid 3, para 1 woman was referred to our unit at 23+2 weeks of gestation presenting with a cyst in the lung of the fetus. She had no past medical history, no operation history, and had not taken any kind of medication, except for oral iron for the current pregnancy. Her family did not have any history of medical illnesses, and she did not smoke nor drink. She had one full-term vaginal delivery two years prior. At the beginning of the current pregnancy, the patient did not have any obstetrical problems. Mid-gestation screening was reported low risk. A 1.8 cm-sized hypoechogenic single cyst in the right lower lobe was detected on ultrasound examination at 21+2 weeks of gestation and was increased to 2.8 cm-size until 23 weeks of gestation. Therefore, she was referred to our hospital at 23+2 weeks of gestation due to a cyst increased in sized in the lung of the fetus.
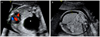
Sonographs revealed a 3 cm-sized hypoechogenic single cyst in the right lower lobe. Displacement of the heart and mediastinum to the left and compression of the heart were seen with normal Doppler images of the umbilical cord and the ductus venosus (Fig. 1A). The patient underwent amniocentesis, and a normal karyotype was detected and no other fetal abnormality was observed. On the basis of the detection of a large and predominant cystlike mass, our tentative prenatal diagnosis was CCAM (type I) [7]. The patient was counseled regarding the etiologies considered and prognosis and informed to the symptoms of polyhydramnios. The mass was followed by serial sonographic examination to determine its rate of growth as well as to detect early signs of fetal problems. There were signs of fetal hydrops on the first visit but no exacerbation was observed on the weekly follow-ups (Fig. 1B). The patient underwent amnioreduction procedure to treat polyhydramnios found at 25+2 weeks of gestation, but amniotic fluid volume continued to rather increase until 32+2 weeks of gestation. Subsequent amnioreductions were performed weekly for polyhydramnios, and then amniotic fluid level was normalized. At 25+2 weeks of gestation, the size of the cyst increased to 4.4 cm. Nearly 30 mL of cystic fluid was aspirated to reduce its size to 2.8 cm, but the cyst recurred to increase in size again to 4.3 cm on the following week. A total of 6 cyst aspirations were performed by 32+2 weeks of gestation. The 4.7 cm-sized hypoechogenic single cyst did not increase in size and was observed until delivery.
At 38+5 weeks' gestation, the mother had a Cesarean delivery to ensure effective care immediately after birth. The 2,792-g male neonate had Apgar scores of 8 and 9 at 1 and 5 minutes, respectively. The baby was admitted to the neonatal intensive care unit for observation. There were no signs of respiratory distress and his neonatal course was uneventful. A chest radiograph showed a huge cystic lesion in the right hemithorax with displaced mediastinal structures such as movement of the heart to left side and atelectatic change of the left lung which was compatible with the prenatal diagnosis of CCAM (type I). A computed tomography (CT) scan on 2 day showed multiple variable sized cystic lesions. The largest was 5.5 cm in diameter, located in the right lung and ground glass opacity in the remaning lungs, suggesting atelectasis (Fig. 2). He had respiratory distress 2 days after birth and was tachypneic without cyanosis. At 3 days of age, a thoracotomy with right lower lobe lobectomy was performed. Biopsy confirmed the diagnosis of CLE with hemorrhage and mild inflammation. Postoperatively the baby developed a left pneumothorax which required drainage. Thereafter, recovery was excellent and the baby was discharged at 4 weeks of age.
Discussion
Congenital lobar emphysema is a rare congenital pulmonary anomaly characterized by air trapping within one or more of the pulmonary lobes with secondary overdistension leading to compression of the remaining lobes and mediastinal shift [1]. The incidence is 1 per 20,000 to 30,000 deliveries, whereas the incidence in utero is unknown [4]. The pathophysiology of CLE consists of disruptions of brochopulmonary development resulting in intrinsic obstruction of bronchus [4]. Intrinsic obstruction involves dysplastic bronchial cartilage or endobronchial lesion with blockage due to mucosal fold, septa, or mucosal plugs [6]. Diffuse bronchial inflammation secondary to congenital cytomegalovirus infection has been reported as a cause of CLE [8]. However, the exact cause of CLE is difficult to determine, and a definitive cause cannot be identified in approximately 50% of cause [6].
Antenatal imaging with sonography has resulted in early identification and serial evaluation of congenital lung lesions. The antenatal diagnosis of lung abnormalities characterized by cyst formation or increased echogenicity include CCAM, pulmonary sequestration, bronchogenic cysts, and, less commonly, congenital lobar emphysema. With CCAM, sonography shows a large cyst (3 to 10 mm in diameter, type I), multiple cyst (0.5 to 2.0 cm in diameter, type II), and the small cystic or solid mass (type III), usually involving one lobe of the lungs [9]. The natural history of fetal CCAMs is variable but most cases of prenatally diagnosed CCAM have good outcome. Prenatal treatment can considered by means of cyst aspiration or thoraco-amniotic shunting and these procedure should be considered before 32 to 33 weeks of gestation. Pulmonary sequestration appears, as a well-defined, solid, and triangular mass and is supplied by an anomalous systemic artery that is usually situated in the lower lobes [10]. The natural history of pulmonary sequestration depends on where they are located, however a considerable number of pulmonary sequestration lesions regress dramatically before birth. Bronchogenic cysts may present as single or, less commonly, as multiple lesions. Their dimensions vary from a few millimeters to greater than 5 cm [11]. For its prognosis and treatment may vary, differential diagnosis is hence needed to diagnose the specific disease in a patient. Prenatal diagnosis of CLE is rarely reported in the literature, because the fetal sonographic features of CLE have not been well described and its prevalence in utero is low. Therefore, in most cases, the diagnosis of CCAM will be suspected, but the correct diagnosis of CLE can only be made postnatally on the basis of histology. Often CLE, CCAM, PS, and bronchogenic cysts are described as one clinical group representing a spectrum of the same disease [12].
Mostly, fetal sonographic features of CLE have a bright echogenic lung without abnormal blood flow. This increased echogenicity is thought to result from excessive lung fluid in the alveoli, and negative intra-thoracic pressure postnatally that results in airway collapse and air trapping leading to the manifestation of CLE [4,6]. Thus far, 3 cases of cystic lesions have been reported (Table 1) [5,6,13]. Antenatal imaging with sonography has shown polyhydramnios and fetal hydrops due to disturbed fetal circulation and impaired swallowing. There may also be a mediastinal shift, leading to fetal impairment. All the lesions mentioned above are predictors of severe respiratory distress or poor outcomes [14]. The antenatal detection of fetal lung echogenicity allows pediastric follow up after birth, which makes possible early diagnosis and managemenst before the infant has symptoms.
Clinically CLE can present with acute symptoms such as tachypnea, respiratory distress, or cyanosis. Simple chest X-ray is usually sufficient for CLE diagnosis, demonstrating lobar hyperinflation, mediastinal shfit and atelectasis of the opposite lung. However if chest radiography is not definitive, a chest CT can be helpful in determining the correct diagnosis. Standard treatment for acute respiratory distress is surgical resection of the affected lung area [3]. Prognosis and survival with surgery are good [15].
There are many reports of spontaneous regression of pulmonary sequestration and CCAM, similar to these lesions, CLE can decrease in size during pregnancy [5]. Therefore, even when they appear to have disappeared, continuing follow-up of prenatal sonographic abnormalities is required.
The prenatal diagnosis of CLE is very rare and the fetal sonographic features of CLE have not been well described. Typically, fetal sonographic features of CLE show a bright echogenic lung and, thus far, only 3 cases have been reported to have cystic lesion. This case reports that CLE should be included in the differential diagnosis of an antenatally detected hypoechoic cystic lung apart from CCAM and pulmonary sequestration.


 XML Download
XML Download