

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Non-Hodgkin's lymphoma (NHL) refers to all malignancies of the lymphoid system with the exception of Hodgkin's lymphoma and is around 90% of all malignant lymphomas [1]. Most patients with NHL present with painless lymphadenopathy, commonly in the cervical or supraclavicular regions [2]. However, extranodal presentation of NHL occurs in 15% to 40% of patients at presentation and varies depending on immune status and geographic differences. The most common extranodal sites are the gastrointestinal tract and nasopharynx. Other common sites include skin, bone, brain, lung, thyroid, salivary glands, and testis [2-6]. Secondary involvement of the adrenal gland with NHL has been reported to occur in up to 25% of patients during the course of disease [2,7]. However, primary adrenal lymphoma (PAL) is a very rare disease and is often accompanied by adrenal insufficiency. The first case of PAL with adrenal insufficiency was reported in 1970 and only ten cases of PAL had been reported until the year 1989 [8]. The prognosis of PAL is known as poor [8-10], but early diagnosis and combination chemotherapy may improve not only the prognosis of lymphoma, but also adrenal function [8].
PAL should be considered in patients with bilateral enlargement of adrenal glands, especially with adrenal insufficiency [8]. We report a case of PAL with primary adrenal insufficiency, which showed early diagnosis and treatment could improve the prognosis.
CASE REPORT
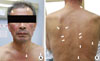
A 54-year old man was hospitalized because of easy fatigability, weight loss of 10 kg and consistent malaise for 6 months. On admission, his vital signs were as follows: body temperature, 36.7℃; pulse rate 113 beats per minute; blood pressure 98/77 mmHg. Physical examination revealed some pigmented spots on his upper chest and face (Fig. 1). His tongue was dry and skin turgor was decreased. Neither lymphadenopathy or hepatosplenomegaly was noted. Ascites and edema were not present.
A complete blood count showed hemoglobin 12.4 g/dL, hematocrit 35.2%, white blood cell count 8320/mL with 58.7% segmented neutrophils, 24% lymphocytes, and 8.8% monocytes, and a platelet count 234,000/mL. The serum sodium, potassium and chloride levels were 128, 4.7, and 91 mEq/L, respectively. The serum β2 microglobulin increased to 3.31 mg/L (normal, 0.0-2.71 mg/L). The serum lactate dehydrogenase level increased to 285 IU/L (normal, 119-247 IU/L). The serum cortisol (8 AM) was 47.85 ng/mL (normal, 70-250) and the plasma adrenocorticotrophic hormone (ACTH) (8 AM) increased to 147.29 pg/mL (normal, 10-60). Twenty four-hour urine vanillylmandelic acid, epinephrine, metanephrine, norepinephrine, and free cortisol were within normal. plasma renin activity increased to 23.53 ng/mL/hr (normal, 0.15-2.23 ng/mL/hr) and aldosterone level was 98.88 pg/mL (normal, 29.9-158.8 pg/mL). We suspect adrenal insufficiency because of hyponatremia, fatigability, weight loss, consistent malaise and hypotension. Clinical suspicion of adrenal insufficiency was confirmed by the finding of a low serum cortisol level of 47.85 ng/mL at 8 AM, a high plasma ACTH level of 147.29 pg/mL at 8 AM, and low cortisol level of 100.14 ng/mL 1 hour after the rapid ACTH stimulation test.
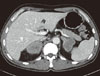
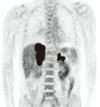
The chest X-ray was normal without evidence of hilar lymphadenopathy. The patient was aged person with prominent weight loss, so we took a computerized tomographic (CT) scan. A CT scan showed large bilateral adrenal gland masses with poor enhancement (Fig. 2). Right adrenal mass invaded S7 of liver and was adjacent to right diaphragm. There were no lymphadenopathy and ascites. Our impressions were lymphoma, metastasis, or adrenal carcinoma. Fludeoxyglucose positron emission tomography (FDG PET)-CT scan was performed to identify the masses and showed huge bilateral adrenal masses with intense FDG uptake and invasion of right adrenal mass into the right hepatic lobe (Fig. 3). These findings are consistent with malignancy. In the rest of the body, there was no increased FDG uptake which suggests malignancy. An ultrasound-guided percutaneous gun biopsy of the right adrenal mass was performed. The gun biopsied specimen showed diffuse proliferation of large atypical cells without forming organoid structures. The tumor cells showed folded nuclei with one or multiple prominent nucleoli and a variable amount of cytoplasm (Fig. 4A). They showed positive reactions with CD20 on immunohistochemical stain (Fig. 4B), but negative reactions with CD3, CD10, and cytokeratin. The final diagnosis was bilateral adrenal diffuse large B cell lymphoma (DLBCL) with adrenal insufficiency, and the clinical staging by the Ann Arbor system was IVB, and international prognostic index was low intermediate risk group.
The patient received adrenal hormone replacement with Cortico® (hydrocortisone) 30 mg daily. He also was administered combination chemotherapy with R-CHOP regimen; rituximab, cyclophosphamide, doxorubicin, vincristine, prednisolone. After 3 cycles of chemotherapy, left adrenal mass was not visible and right adrenal mass decreased in size from 6.4 cm to 3.7 cm by CT scan. He received total 8 cycles of chemotherapy, because PET scan showed mild hypermetabolism on right adrenal gland after sixth cycle of chemotherapy. After 8 cycles of chemotherapy, right adrenal mass further decreased in size from 3.7 cm to 3.5 cm by CT scan and there were no increased FDG uptake by PET scan (Fig. 5). He had taken a prednisolone 10 mg per day for 6 months. Clinically adrenal insufficiency was also improved with disease remission, so the dose of steroid replacement was tapered off. This patient is continued to be followed up without any evidence of recurrence. Furthermore, he did not have any signs or symptoms of adrenal insufficiency for 1 year.
DISCUSSION
Adrenal glands are common metastatic site of primary cancers, such as breast, lung, lymphoma, melanoma, leukemia, renal cell and ovarian cancers [11]. In contrast to primary tumors, many metastases tend to invade both adrenal glands. Primary malignant lymphomas arising in endocrine organs are rare with representing only 3% of extranodal lymphomas [8]. They are almost confined to the thyroid gland [5,12] and primary lymphoma originated from adrenal is extremely rare. The first case of PAL with adrenal insufficiency was reported in 1970 and only ten cases of PAL had been reported until the year 1989. The development of imaging techniques and immunostaining procedures makes the diagnosis of PAL easier and so, about 120 cases of PAL have been reported in the literature [9].
Adrenal insufficiency is rare because it becomes apparent when approximately 90% of adrenal cortex is destructed [13]. And most of adrenal insufficiency are believed to be associated with atrophy of adrenal glands, secondary to autoimmune mechanisms [14]. Approximately 50% of patients with primary adrenal lymphoma present with symptoms of adrenal insufficiency [9]. Our patient also showed typical symptoms and sings of adrenal failure. In this patient, dehydration accompanied with by adrenal insufficiency may cause to increase renin activity. Human adrenal glands contain no lymphoid tissue, so PAL is believed to arise from previous autoimmune adrenalitis, which explain why adrenal insufficiency is common in PAL. The symptoms of adrenal insufficiency are general weakness, fatigue, weight loss, hyperpigmentation, hypotension, fever, and gastrointestinal symptoms such as anorexia, nausea, vomiting and dyspepsia [9]. Among disease symptoms, hyperpigmentation is usually present with long term adrenal insufficiency. Because DLBCL is aggressive disease, most PAL patients with adrenal insufficiency are not present with hyperpigmentation. However, our patient had some pigmented spots on his upper chest and face (Fig. 1), which mean chronic progression of the disease. Primary adrenal insufficiency can be confirmed by low basal serum cortisol level, high ACTH level, and inadequate adrenal response to the rapid ACTH stimulations [14].
In PAL, men are more frequently affected than women, with a ratio of 3:1. The mean age at the time of diagnosis is 68 years (range 39-89). More than two-third of PAL are bilateral. Most common histopathology is diffuse large cell lymphoma of B cell lineage [9]. These malignant cells express CD45, and antigens specific to B lymphocytes, such as CD19, CD20, and CD79a [15]. About 10% of de novo DLBCL cases express CD5 and CD5-positive DLBCL is more common in older women, with extranodal involvement and associated with a shorter survival [15]. T cell lymphoma, expressing antigens specific to T lymphocytes, such as CD3, CD5, and CD7 [16] occupies less than 10% of PAL. In our case, the tumor cells showed positive reactions with CD20 on immunohistochemical stain, but negative reactions with CD3 and CD10. Less common histopathology includes small-cell lymphoma, mixed small- and large-cell lymphoma, undifferentiated lymphoma, anaplastic large-cell and follicular lymphoma [9]. On CT scan, lymphomas usually show complex masses with variable density and sometimes accompanied by necrosis and hemorrhage, but calcification is uncommon. The contour of the involved adrenal gland may be preserved. On MRI scan, lymphomas show low signal intensity on T1-weighted images and heterogeneous high signal intensity on T2-weighted images. However, CT and MRI finding of adrenal lymphoma are not specific [17]. In our case, CT scan showed large bilateral adrenal gland masses with poor enhancement, and lymphoma, metastasis or adrenal carcinoma were included in the differential diagnosis. FDG PET scan is useful for diagnosis of malignancy. And if malignancy is diagnosed, FDG PET can be useful for detecting tumor recurrences and response to treatment [8]. Percutaneous needle biopsy under CT or US guidance has been reported as a safe and accurate diagnostic tool. Before a biopsy in patient with adrenal masses, we must exclude pheochromocytoma to avoid a hypertensive crisis.
The prognosis of PAL is known as poor, but early diagnosis of the disease and combination chemotherapy may improve the prognosis. Several poor prognostic factors of PAL are old age, adrenal insufficiency, large tumor size, elevated serum LDH level and expression of CD5 [18,19]. The cause of death is severe infection, pulmonary embolism, and tumor recurrence [20]. One study reported that mean overall-survival rate is 15.3 months; 34 months of the patients who achieve a partial or complete remission after chemotherapy and 3.6 months of the patients who have no response to chemotherapy [8]. The longest survival was 50 months in cases of treating with CHOP chemotherapy [13]. The treatment of PAL is not established, but recent studies show that chemotherapy including CHOP can extend survival. The regimens of chemotherapy include CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), R-CHOP (adding rituximab to CHOP), CHO (cyclophosphamide, doxorubicin, vincristine), CVP (cyclophosphamide, vincristine, and prednisone) or MACOP (cyclophosphamide, doxorubicin, prednisone, methotrexate, bleomycin) [9]. Surgical resection only is not recommended for PAL treatment because of a poor outcome. The role of radiation therapy on PAL is unknown. But in some cases, the patients with PAL had radiation therapy with chemotherapy [9]. CNS prophylaxis with intrathecal methotrexate is also considered in patients with high risk of CNS involvement. Risk factors for CNS involvement are elevated serum LDH level, high/intermediate or high International Prognostic Index and involvement of more than one extranodal site including bone marrow [9].
In this case, the patient had taken abdomen ultrasonography and CT at first visit of our hospital, and diagnosed of NHL by percutaneous gun biopsy. After pathologic diagnosis, he received combination chemotherapy without delay. After 8-cycle of chemotherapy, he achieved complete remission clinically. Primary adrenal lymphoma, although a rare disease, should be considered in patients with bilateral enlargement of adrenal glands, especially with adrenal insufficiency. And early diagnosis can lead to better prognosis by early treatment.


 XML Download
XML Download